Case preview: Thalassemia - tài liệu tham khảo môn sinh học - Đại học Y dược Thành phố Hồ Chí Minh
Thalassemia(s) là một nhóm bệnh di truyền trong đó đột biến gene liên quan đến quá trình tổng hợp chuỗi globin của Hemoglobin (Hb) dẫn đến mất cân bằng tương đối giữa các chuỗi globin. Phổ biến nhất là đột biến gen ɑ hay βglobin gây nên bệnh ɑ-thalassemia hoặc βthalassemia. Ngoài ra đột biến còn có thể tác động đến một số gen tương đồng trong cụm gen ɑ hay β như gen γ, gene δ,… gây nên bệnh δthalassemia và γ-thalassemia nhưng hiếm gặp và triệu chứng lâm sàng không đáng kể..... Tài liệu tham khảo bổ ích kèm bài tập ứng dụng, mời các bạn đón đọc
Môn: Sinh di truyền 10 tài liệu
Trường: Đại học Y Dược Thành phố Hồ Chí Minh 379 tài liệu
Tác giả:

















Preview text:
lOMoAR cPSD| 51038363 ĐÁI THÁO NHẠT
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363 MỤC LỤC
TỔNG QUAN .......................................................................................................................................... 2
Giới thiệu chung ...................................................................................................................................... 2
Các gen globin .......................................................................................................................................... 2
Sắp xếp các gen globin trên NST ........................................................................................................ 3
Kiểm soát biểu hiện gen ....................................................................................................................... 3
Chẩn đoán – Các chỉ số huyết học ...................................................................................................... 4
BỆNH β-THALASSEMIA ................................................................................................................... 5
Tổng quan ................................................................................................................................................. 5
Khiếm khuyết gene gây ra β-thalassemia ...................................................................................... 6
Cơ chế bệnh sinh cấp độ tế bào của bệnh β-thalassemia.......................................................... 7
BIỂU HIỆN LÂM SÀNG CỦA BỆNH β-THALASSEMIA .......................................................... 8
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 1
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
Thể dị hợp tử β-thalassemia .............................................................................................................. 8
β-thalassemia thể nặng (bệnh thiếu máu Cooley) ...................................................................... 9
β-thalassemia thể trung gian........................................................................................................... 11
Tương tác của β-thalassemia với các biến thể cấu trúc Hb phổ biến ................................ 11
BỆNH α-THALASSEMIA ................................................................................................................ 12
Tổng quan bệnh ................................................................................................................................... 12
Các dạng bệnh và triệu chứng lâm sàng ...................................................................................... 14
CÂU HỎI ÔN TẬP ............................................................................................................................. 15
ĐÁP ÁN ................................................................................................................................................ 16
TÀI LIỆU THAM KHẢO .................................................................................................................. 17
Case Preview: Module 1 – Từ Phân Tử Đến Tế Bào THALASSEMIA
Tái bản lần 2: Nguyễn Văn Trung, Nguyễn Đăng Nhân và cộng sự
Cố vấn: Phùng Gia Bảo, Nguyễn Huyền Ngọc Mai 1/2024 TỔNG QUAN Giới thiệu chung
Thalassemia(s) là một nhóm bệnh di truyền
trong đó đột biến gene liên quan đến quá trình
tổng hợp chuỗi globin của Hemoglobin (Hb) dẫn
đến mất cân bằng tương đối giữa các chuỗi
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 2
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
globin. Phổ biến nhất là đột biến gen ɑ hay Các gen globin
βglobin gây nên bệnh ɑ-thalassemia hoặc
Thành phần Hb
βthalassemia. Ngoài ra đột biến còn có thể tác
động đến một số gen tương đồng trong cụm gen
Hb trưởng thành (adult Hb) là một hỗn hợp
ɑ hay β như gen γ, gene δ,… gây nên bệnh
protein không đồng nhất (heterogeneous) bao
δthalassemia và γ-thalassemia nhưng hiếm gặp
gồm thành phần chính là HbA và thành phần
và triệu chứng lâm sàng không đáng kể (Xem
phụ là HbA2 - chiếm khoảng 2,5%. Trong Hb Hình 1).
bào thai (fetus Hb), Hb chính là Hb F. Mỗi đại
phân tử Hb được cấu thành từ hai cặp tiểu đơn
vị globin khác biệt nhau về cấu trúc. Ngoại trừ
một số Hb phôi sớm (embryo Hb), tất cả các đại
phân tử Hb ở người bình thường đều có một
cặp αglobin. Ở HbA, 2 chuỗi α kết hợp với 2
chuỗi β (α2β2); ở HbA2 có 2 chuỗi δ (α2δ2); và ở
HbF có 2 chuỗi γ (α2γ2).
Đặc biệt ở Hb bào thai, sự không đồng nhất
trong thành phần huyết sắc tố còn đáng kể hơn
nữa. HbF là hỗn hợp của hai loại phân tử có
Hình 1: Những dạng khác nhau của tập hợp bệnh công thức α Thalassemias.
2γ2136Gly và α2γ2136Ala. Chuỗi γ chứa
glycine ở vị trí 136 được kí hiệu là chuỗi Gγ.
Mặc dù đa dạng về nguyên nhân và mức độ
Chuỗi γ chứa alanine được gọi là chuỗi Aγ. Khi
nghiêm trọng, bệnh thalassemia có thể được chia
sinh, tỷ lệ số lượng phân tử Hb chứa chuỗi Gγ
thành 2 dạng là: thalassemia phụ thuộc truyền
so với Hb chứa chuỗi Aγ xấp xỉ 3:1. Trước tuần
máu (tranfusion-dependant thalassemia, TDT)
thứ 8 của bào thai, trong phôi hiện diện ba loại
và thalassemia không phụ thuộc truyền máu
Hb - Gower 1 (ξ2ε2), Gower 2 (α2ε2) và Portland
(non-tranfusion-dependant thalassemia, NTDT).
1 (ξ2γ2). Chuỗi ξ ở phôi tương ứng với chuỗi α
Bệnh thalassemia thu hút sự quan tâm và chú ý
ở người trưởng thành, chuỗi ε tương ứng với
trên toàn thế giới do mức độ phổ biến rất lớn của
chuỗi β, γ, δ. Trong quá trình phôi phát triển
người mang gen bệnh và tầm quan trọng trên lâm
thành thai, khi bánh nhau được thành lập, xảy
sàng của bệnh cũng như gánh nặng kinh tế trong
ra sự chuyển đổi có trật tự từ sản xuất chuỗi ξ
việc điều trị suốt đời cho những bệnh nhân (BN)
sang chuỗi α và từ chuỗi ε sang chuỗi γ; theo
TDT. Ngoài ra, sự phát triển của sinh học phân tử
sau là sản xuất chuỗi β và δ sau khi sinh.
đã cung cấp một bức tranh rõ ràng hơn về cơ chế
bệnh sinh của bệnh thalassemia; và từ đó rút ra
Sắp xếp các gen globin trên NST
được những vấn đề nền tảng của sinh học; đặc
(Xem Hình 2) Thể hiện sơ đồ bố trí các gen
biệt là sự điều hoà gen có tính đặc hiệu mô
globin ở người. Có 2 domain gen α-globin nằm
(tissue-specific) và có tính đặc hiệu theo giai
nối tiếp trên NST 16 (ký hiệu là α2 và α1), phía
đoạn phát triển của cơ thể (development-
downstream so với 2 domain gen phôi sớm specific).
giống-α (α-like) được gọi là zeta (ζ, cụ thể là ζ2 và
ζ1). Tính tương đồng cao giữa các domain gen α-
globin dẫn đến thường xuyên xảy ra các hiện
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 3
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
tượng trao đổi chéo không đồng đều khi giảm
thể. Trong suốt thai kỳ, cả hai NST phối hợp
phân; đây là cơ sở cho các đột biến mất đoạn
tổng hợp các chuỗi globin, đảm bảo cho sự sản
domain α gây ra bệnh α-thalassemia.
xuất theo tuần tự và có trật tự các tetramer Hb
chức năng. Trong quá trình biến đổi từ phôi
sang thai sang sơ sinh, các gene globin trên cả
hai NST được “đọc” (được lựa chọn phiên mã)
tuần tự từ trái sang phải (đầu 5′ đến đầu 3′).
Trong tháng đầu tiên của thai kỳ, Hb phôi sớm
(ζ2ε2, α2ε2, ζ2γ2) được hình thành trong các tế
bào (TB) dòng hồng cầu chủ yếu tại túi noãn
hoàng. Trong khoảng thời gian còn lại của thai
kỳ, các vị trí xảy ra sự tạo hồng cầu
Hình 2: Bố trí họ gen α-globin trên NST 16 và họ
(erythropoiesis) dần dần dịch chuyển từ gan và
gen β-globin trên NST 11. IVS, vùng ngăn cách (hay lách đến tủy xương.
còn gọi là intron); Ba vùng exon của các gen globin
HbF (α2γ2) là Hb chính trong hồng cầu của bào
được trình bày trong hình có màu xanh dương nhạt.
thai. Ngay trước khi sinh, xảy ra một sự chuyển
đổi lớn từ tổng hợp chuỗi γ-globin sang
Cụm (cluster) gen giống-β (β-like) globin nằm
βglobin, và hoàn tất khi trẻ được 6 đến 8 tháng
trên NST 11 gồm 6 domain, trong đó có 1 giảgen
tuổi sau sinh. Kể từ đó, hơn 95% Hb trong hồng
(pseudogene) là ψβ, có cấu trúc rất tương đồng
cầu bình thường là HbA (α2β2). Cơ sở sinh học
với các gen globin nhưng không hoạt động và 5
phân tử của sự chuyển đổi này là một trong
gen khác. Giống như cụm gen α-like globin,
những vấn đề quan trọng nhất trong nghiên
epsilon (ε) là gen nằm xa nhất về phía đầu 5′
cứu huyết học, và nó tác dụng đến cơ chế bệnh
(upstream) của cụm gen β-globin và chỉ được
sinh và điều trị β-thalassemia.
biểu hiện ở đầu kỳ phôi bào. Theo sau là hai gen
gamma (Gγ và Aγ) nối tiếp nhau, chúng mã hoá
cho các chuỗi globin γ cấu thành nên Hb bào thai
(HbF, α2γ2). Sản phẩm mã hoá của gen delta (δ)
tạo thành một dạng Hb thiểu số là HbA2 (α2δ2).
Dù không thực hiện chức năng đáng kể nào, xét
nghiệm nồng độ HbA2 rất hữu ích trong chẩn
đoán bệnh thalassemia. Thành viên nằm xa
Hình 3: Vị trí xảy ra sự tạo hồng cầu và diễn biến
nhất về phía đầu 3′ (downstream) của gen
tổng hợp chuỗi globin.
βglobin là gen β, tổng hợp protein β-globin kết
hợp với α-globin để tạo thành HbA (α
Phiên mã, xử lý hậu phiên mã và dịch mã 2β2), dạng
Hb chính ở hồng cầu của người trưởng thành.
Trong suốt quá trình trưởng thành, các TB dòng
Kiểm soát biểu hiện gen
hồng cầu ngày càng được biệt hoá cho việc sản
xuất Hb. Xa về phía upstream của các gen globin
Phụ thuộc vào giai đoạn cơ thể phát triển
là các yếu tố điều hòa, được gọi là vùng kiểm soát
(Xem Hình 3) Mức độ biểu hiện của các gen
locus (locus control region). Tại đó yếu tố phiên
globin tuỳ thuộc vào giai đoạn phát triển của cơ
mã đặc hiệu dòng hồng cầu (erythroidspecific
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 4
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
transcription factor) phối hợp liên kết với yếu tố
thành các tiểu đơn vị (subunit) - cấu phần cơ sở
đáp ứng trên DNA để đảm bảo diễn ra sự phiên
của Hb heterotetramer (hetero: dị hợp), ví dụ
mã tạo ra các α- và β- (hoặc γ-) globin RNA có tính
như Hb A (α2β2). Nếu xảy ra sự mất cân bằng
đặc hiệu theo mô, diễn ra ở tần suất cao và trùng
tổng hợp giữa các chuỗi α- và β-globin, ví dụ
khớp lẫn nhau về mặt thời gian và số lượng.
đột biến bất hoạt biểu hiện gen α và dẫn đến dư
Tương tự như các mRNA sơ khai khác, mRNA
thừa chuỗi β-globin hoặc ngược lại, mức độ dư
globin phải trải qua quá trình xử lý thành mRNA
thừa của chuỗi globin tự do sẽ tạo điều kiện
trưởng thành và được vận chuyển từ nhân đến TB
hình thành các thể vùi (kết tủa) và gây tổn
chất để diễn ra sự dịch mã (tổng hợp protein). Tất
thương đến TB – bệnh thalassemia. (Xem Hình
cả các gen globin đều có ba exon và hai intron.
5) Mất cân bằng tổng hợp chuỗi globin qua tỷ
(Xem Hình 4) Minh hoạ quá trình xử lý mRNA sơ
lệ β-/αglobin trong các dạng bệnh thalassemia.
khai bao gồm: cắt bỏ tuần tự hai đoạn intron và ghép nối các đoạn exon
(splicing), đóng khoá đầu 5’ (capping) của chuỗi
mRNA nhằm tăng hiệu quả phiên mã, phân cắt
(cleavage) và polyadenyl-hoá vùng không dịch
mã (untranslated region, UT) tại đầu 3’ nhằm
tăng tính ổn định của mARN. Đột biến xảy ra tại
các vị trí quan trọng liên quan đến sự cắt nối,
đóng khoá và polyadenyl-hoá có thể dẫn đến
khiếm khuyết trong quá trình tổng hợp globin và gây nên bệnh thalassemia.
Hình 5: Tỷ lệ tổng hợp sinh học chuỗi β-/α-globin
trong các dạng bệnh thalassemia.
Chẩn đoán phân biệt các chỉ số huyết học
Thiếu máu thiếu sắt
Thiếu máu thiếu sắt xảy ra khi không có đủ
lượng sắt trong cơ thể để tạo ra hemoglobin.
Thiếu sắt là nguyên nhân gây ra nhiều chứng
Hình 4: Quá trình xử lý mRNA sơ khai được phiên
bệnh nguy hiểm cho trẻ em, phụ nữ và người
mã từ gen β-globin.
cao tuổi, đặc biệt là các chứng bệnh liên quan
Khi các TB tiền thân dòng hồng cầu trưởng thành
đến hệ tim mạch, hô hấp. β-thalassemia
hoàn toàn, chúng dành hơn 95% sản lượng tổng
hợp protein cho sản xuất chuỗi globin; và
tổng hợp nhóm ngoại hem
(protoporphyrin IX + Fe2+) cũng tăng tương
ứng. Ở các giai đoạn trưởng thành của hồng cầu
sau khi được giải phóng khỏi phức hợp
polyribosome, globin kết hợp với hem để tạo
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 5
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
β-globin (β+) hoặc không có sự tổng hợp
ra đột biến mất đoạn cis (trên cùng NST) hoặc βglobin (β
trans (khác NST). Kiểu gen bình thường là
0). Bệnh này thường phổ biến ở người
αα/αα. Ngược lại với thiếu máu do thiếu sắt,
gốc Địa Trung Hải. α- thalassemia
alpha-thalassemia thường làm tăng số lượng
Mất đoạn gen α-globin trên nhiễm sắc thể 16, dẫn
hồng cầu RBC, nhưng các hồng cầu này lại nhỏ
đến giảm sản xuất hemoglobin. Có thể diễn
và chứa ít hemoglobin hơn bình thường. Tần
suất mắc bệnh cao ở người gốc Á và Phi. Xét nghiệm Thiếu sắt β-thalassemia α- thalassemia
MCV (thể tích trung bình hồng cầu) Thấp Thấp Thấp
RDW (độ phân bố hồng cầu) Cao Bình thường Bình thường; thỉnh thoảng cao Ferritin Thấp Bình thường Bình thường > 13 < 13 < 13 Chỉ số Mentzer cho trẻ em (MCV/RBC) Điện di huyết sắc tố
Bình thường (có HbA2 tăng HbA Người lớn: đa số (Hb electrophoresis) thể có HbA2 giảm giảm bình thường Trẻ khi thiếu máu HbF có thể tăng sơ sinh: có thể có nặng) HbH hoặc Hb Bart's
Bảng : So sánh thiếu máu thiếu sắt, β-thalassemia, α- thalassemia qua các chỉ số cận lâm sàng. (Nguồn:
Muncie HL, Campbell J. Alpha and beta thalassemia. American Family Physician. 2009;80(4):339-
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 6
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
Là bệnh xảy ra do đột biến điểm ở vị trí cắt nối
βthalassemia, một người nếu cùng lúc nhận hai
(splicing sites) hoặc trình tự Kozak (promoter)
đột biến khác nhau từ bố và mẹ thì mang KG dị
trên nhiễm sắc thể số 11, dẫn đến giảm tổng
hợp tử phức hợp (compound heterozygotes). hợp 344.
Nếu KG đồng hợp tử hoặc dị hợp tử phức hợp
https://pubmed.ncbi.nlm.nih.gov/19678601)
biểu hiện ra bệnh nặng thì được xác định là mắc
bệnh β-thalassemia thể nặng (β-thalassemia BỆNH β-THALASSEMIA
major, hay bệnh thiếu máu Cooley); ngược lại
nếu biểu hiện lâm sàng nhẹ hơn, thì được xác
định là mắc bệnh β-thalassemia thể trung gian Tổng quan (β-thalassemia intermedia).
Như đã trình bày ở trên, NST 11 chỉ chứa một
Hơn 100 triệu người trên thế giới mang KG dị
domain gen β-globin đơn lẻ. Những người thừa
hợp tử chứa một đột biến β-thalassemia: gần
hưởng đột biến β-thalassemia từ bố hoặc mẹ và
hai phần ba trong số đó đến từ châu Á và phần
thừa hưởng gen β-globin bình thường từ người
còn lại được phân bổ giữa các khu vực châu Phi,
còn lại thì mang kiểu gen (KG) dị hợp tử
châu Âu và châu Mỹ; tập trung ở khu vực nhiệt
(heterozygotes), thường được biểu hiện ra kiểu
đới. Theo nghiên cứu, KG dị hợp tử β-
hình là người lành mang gen bệnh (healthy
thalassemia được bảo vệ chống lại bệnh sốt rét
carrier), hay β-thalassemia trait, hay β-
do KST Plasmodium falciparum ở trẻ sơ sinh
thalassemia thể nhẹ (β-thalassemia minor).
(infantile falciparum malaria) – bệnh này rất
Những người thừa hưởng cùng lúc hai đột biến
nguy hiểm và thường gây tử vong. Nguyên
β-thalassemia giống nhau từ cả bố và mẹ thì
nhân dẫn đến tần số gen β-thalassemia cao ở
mang KG đồng hợp tử (homozygotes). Bởi vì
quần thể nơi bệnh sốt rét lưu hành là nhờ vào
nhiều loại đột biến có thể làm phát sinh bệnh
việc hồng cầu nhỏ (hậu quả của bệnh
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 7
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
thalassemia) tăng tỉ lệ S/V khiến cho cấu trúc
khởi đầu đầu 5′ hoặc ở vị trí 3′ trong RNA β-
của hồng cầu trở nên bền vững hơn, KST sốt rét
globin, tại đó RNA được phân cắt trước khi
khó có thể phá vỡ hồng cầu để thoát ra ngoài. polyadenyl-hóa.
Đây chính là một hình thức của chọn lọc tự nhiên.
Khiếm khuyết gene gây ra β-thalassemia
Bệnh β-thalassemia phát sinh từ các đột biến
khác nhau liên quan đến vùng promoter, trình tự
mã hóa, ranh giới intron-exon và vị trí
polyadenyl-hoá của gen β-globin (Xem Hình 6).
Các alen đột biến được chia thành hai nhóm: β0 –
mất hoàn toàn khả năng tổng hợp β-globin, và β+
- chỉ tổng hợp được một lượng nhỏ protein β- globin bình thường.
Phần lớn các alen β0-thalassemia liên quan đến
đột biến điểm thay thế nucleotide trong vùng mã
hóa cho chuỗi polypeptide β tạo ra codon kết
thúc sớm, hoặc đột biến mất đoạn/lặp đoạn nhỏ
làm cho lệch khung đọc của mARN - đột biến dịch
khung. Trong cả hai trường hợp, thu được chuỗi
polypeptite bị cắt ngắn, mất chức năng tổng hợp
β-globin. Một số cơ chế di truyền khác ít phổ biến
hơn tạo ra alen β0 bao gồm các đột biến mất đoạn
và thay thế tại điểm giao ghép nối (splice
Hình 6: Cơ chế đột biến cấp độ sinh học phân tử junction).
gây bệnh β-thalassemia.
Trong alen β+-thalassemia, gen khiếm khuyết
Thalassemia là bệnh chủ yếu di truyền lặn trên
vẫn cho phép sản xuất β-globin bình thường,
NST thường (Xem Hình 7), có thể phân biệt
nhưng số lượng giảm rõ rệt do gen bị giảm chức đơn giản:
năng. Loại alen này thường là do một đột biến
• Thể nhẹ: β0/ β hoặc β+/ β
điểm thay thế nucleotide làm tạo ra vị trí ghép
• Thể trung gian (intermedia): β+/ β+
nối mới/sai hoặc làm giảm hiệu quả của vị trí hoặc β+/ β0
ghép nối bình thường. Trong cả hai trường hợp, •
một số hoạt động ghép nối bình thường vẫn xảy
Thể nặng (major): β0/ β0
ra và tạo ra một số phân tử β-globin bình thường,
song số lượng rất ít. Nếu quá trình xử lý mRNA
Hình 7: Di truyền lặn trên NST thường ở β-
sơ khai hậu phiên mã sử dụng các vị trí ghép nối thalassemia.
thay thế, sẽ tạo ra một mRNA trưởng thành vô
nghĩa và không thể tổng hợp được sản phẩm
protein ổn định, hữu ích. Một dạng hiếm gặp hơn
của alen β+-thalassemia là do đột biến ở vùng
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 8
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
Cơ chế bệnh sinh cấp độ tế bào của bệnh β-thalassemia
Hình 8: Quá trình tổng hợp ra các tiểu đơn vị Hb
Trong thành phần của HbF và HbA
và Hb tetramer trong bệnh β-thalassemia. 2, không có
chứa chuỗi β-globin nên không bị ảnh hưởng bởi (2)
Các chuỗi α-globin sẽ tổ hợp lại thành
bệnh β-thalassemia do đó bệnh không biểu hiện
homotetramer α4 không hòa tan được (có tính
ở giai đoạn phôi thai và mới sanh. β-thalassemia
tan kém). Việc này dẫn đến những chuỗi dư có 3 cơ chế chính:
thừa kết tủa và lắng đọng trong tế bào tiền thân •
Tạo hồng cầu không hiệu quả với sự phá
hồng cầu tạo thành những thể vùi, gây ra sự
hủy trong tủy xương với các tỷ lệ khác nhau của
phá hủy tế bào tiền thân hồng cầu trong tủy
dòng tế bào tiền thân hồng cầu đang phát triển.
xương và tạo hồng cầu không hiệu quả. Thể ẩn
và chuỗi α tự do sẽ làm các TB tiền thân dòng •
Tán huyết do hủy hồng cầu trưởng thành
hồng cầu bị phá hủy sớm trong tủy xương gây
có chứa thể vùi của chuỗi α
thiếu máu sớm tại tủy (tán huyết nội tủy) – đặc •
Hồng cầu nhỏ và nhược sắc do giảm tổng
biệt xảy ra nghiêm trọng trong thể nặng. β- hợp hemoglobin chung.
thalassemia dị hợp tử cũng có sự tổng hợp (1)
Vì lượng β-globin tạo ra không đủ đáp
chuỗi globin không cân bằng, nhưng độ dư
ứng nên xảy ra thiếu hụt Hb trong hồng cầu. Do
thừa chuỗi ⍺ thì ít hơn nhiều và có thể được giải
đó, nồng độ Hb trung bình trong hồng cầu
quyết bởi enzymes của tế bào tiền thân hồng
(mean cell Hb, MCH) và thể tích trung bình của
cầu (hệ thống ubiquitinproteasome).
hồng cầu (mean cell volume, MCV) đều bị giảm. (3)
Ngoài ra, thời gian sống sót của các
Sự phá hủy hàng loạt TB dòng hồng cầu ở BN
hồng cầu trưởng thành lưu thông trong máu
được lý giải là do sự mất cân bằng chuỗi protein.
cũng giảm đi một phần do chuỗi α dư thừa có
Như trong Hình 5, tỷ lệ chuỗi β/α được tổng hợp
heme bị oxyhóa hình thành hemichromes, các
là khoảng 0,5 ở thể dị hợp tử; và chỉ còn 0,1 ở thể
hemichromes chứa sắt sẽ thành chất Oxy phản
đồng hợp tử hoặc dị hợp tử phức hợp, nghĩa là
ứng (reactive oxygen species, ROS) sẽ oxy-hóa
BN dư thừa rất nhiều chuỗi α-globin. (Xem Hình
các protein trên màng hồng cầu gây tổn thương
8) Ngoài ra do phản ứng sinh tồn nên có sự tăng
màng hồng cầu. Khi đó đại thực báo sẽ bị thu
tổng hợp chuỗi γ-globin dẫn đến nhiều chuỗi α-
hút và đến phá huỷ trong hệ võng nội mô gồm
globin kết hợp với γ-globin để tạo thành HbF.
tủy, gan và lách; tăng phá hủy hồng cầu bị tổn
HbF chiếm ưu thế trong hồng cầu của BN mắc
thương ở đây (tán huyết tại hệ võng nội mô) -
bệnh β-thalassemia thể nặng.
góp phần vào tình trạng gan, lách to. Những bất
thường này xuất hiện với mức độ nhẹ ở thể dị
hợp tử và với mức độ nghiêm trọng rõ rệt ở thể
đồng hợp tử hoặc dị hợp tử phức hợp.
Ở những BN thiếu máu trầm trọng, sự tăng tiết
hormone erythropoietin EPO (hormone tăng sinh
hồng cầu) bù trừ sẽ kích thích đáng kể quá trình
tạo hồng cầu trong các khoang tủy của tất cả các
xương tạo máu, cũng như ở các vị trí tạo máu
ngoại tủy như gan và lách, dẫn đến các xương tạo
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 9
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
máu có tủy xương ngày càng rộng ra còn vỏ
nồng độ Hb bình thường, nhưng nhiều người bị
xương ngày càng mỏng lại. Kết quả là, BN bị biến
thiếu máu nhẹ. Tất cả những người này đều có
dạng xương như u trán, u chẩm và u đỉnh, xương
hồng cầu nhỏ nhược sắc. Một số có nồng độ
hàm trên nhô ra, sống mũi dẹt, dễ gãy xương vì vỏ
bilirubin không liên hợp tăng phản ánh sự tăng
xương mỏng... (điển hình là “gương mặt
phá hủy TB dòng hồng cầu. Ở một số ít KG dị thalassemia”).
hợp tử, lách to nhẹ. β-thalassemia trait thường
Sự tạo hồng cầu không hiệu quả cộng thêm với
phát hiện tình cờ khi làm xét nghiệm tổng phân
suy giảm thời gian sống sót của hồng cầu trong
tích TB máu ngoại vi bằng máy đếm laser tự
máu lưu thông sẽ biểu hiện thành tình trạng
động, chẩn đoán được tiến hành bằng cách: (1)
thiếu máu. Vì tình trạng thiếu máu trên làm
Đầu tiên loại trừ tình trạng thiếu máu hồng cầu
hepcidin (hormone điều hòa hấp thu sắt trong cơ
nhỏ nhược sắc khác như thiếu máu thiếu sắt,…;
thể, đặc biệt qua đường tiêu hóa) giảm, do đó
(2) và sau đó chứng minh sự gia tăng % HbA2
tăng hấp thu sắt từ đường tiêu hóa vào máu phục
và HbF trong máu bằng phương pháp điện di.
vụ cho tủy tăng tạo hồng cầu. Nếu tình trạng
Trong một phần nhỏ BN, họ có kiểu hình của
thiếu máu nặng nề dẫn đến sự quá tải sắt, có thể
βthalassemia với một alen trong đó mất cả hai
bộc lộ ra ngoài bằng dấu hiệu xạm da; nặng nề
domain gene δ và β. Họ có nồng độ HbA2 thấp
hơn có thể tổn thương lên các tuyến nội tiết như
hoặc bình thường và HbF tăng.
tuyến yên, tuyến giáp, tuyến tuyến tụy, tuyến
sinh dục hay xơ gan ứ sắt, tổn thương lên cơ tim.
Khác nhau cơ bản của cơ chế bệnh sinh của α và
ꞵ-thalassemia chính là việc chuỗi ꞵ hay γ tạo
nên homotetramer. homotetramer α4 khó tan
trong nước trong khi homotetramer γ4 và ꞵ4 thì
ngược lại, do đó chúng không tạo kết tủa và gây
rối loạn trong tổng hợp hồng cầu. Vì vậy, ⍺-
thalassemia không đặc trưng bởi sự tạo hồng
Hình 10: Phết (tiêu bản) máu của một BN mắc
cầu không hiệu quả như ꞵ-thalassemia.
bệnh β-thalassemia thể nhẹ.
Phương pháp điều trị
Hình 9: Tóm tắt cơ chế bệnh sinh cấp độ TB của bệnh β-thalassemia.
Nhìn chung, BN mắc β-thalassemia trait hầu như
không cần điều trị, song cần được đảm bảo rằng
tình trạng này là lành tính và không gây ra vấn
BIỂU HIỆN LÂM SÀNG CỦA BỆNH
đề về sức khỏe. Trước khi sinh con, các bậc cha β-THALASSEMIA
mẹ tương lai nên được tư vấn di truyền về nguy
cơ sinh con bị β-thalassemia thể trung bình đến
Thể dị hợp tử β-thalassemia
nặng nếu cả hai vợ chồng đều là β-thalassemia trait (KG dị hợp tử).
Triệu chứng lâm sàng
Những người mang β-thalassemia KG dị hợp tử
(trait) gần như không biểu hiện triệu chứng và
có tuổi thọ bình thường. Mặc dù hầu hết đều có
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 10
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
β-thalassemia thể nặng (bệnh thiếu máu Cooley)
Hình 11: Phim X quang chụp các xương dài phía
Triệu chứng lâm sàng
xa ở cánh tay và chân của trẻ mắc bệnh
thalassemia thể nặng. Xét nghiệm và chẩn
BN có biểu hiện thiếu máu nặng và thường không đoán
sống sót đến tuổi trưởng thành. Như trong Hình
9, kích thích sản sinh hồng cầu mạnh và kéo dài
Thalassemia thể nặng thường khởi phát vào
qua trung gian erythropoietin dẫn đến sự tạo
khoảng 6 tháng tuổi sau sinh khi chuyển dịch
hồng cầu ngoài tủy và gan, lách to. Hiện tượng
hoàn toàn từ tổng hợp chuỗi γ-globin sang
quá tải sắt tiến triển do tăng cường hấp thu sắt
βglobin. Các nguyên hồng cầu xuất hiện trong
từ đường tiêu hóa, cùng với sự tích tụ sắt sau khi
máu ngoại vi, và số lượng của chúng tăng lên rõ
điều trị truyền máu. BN da trắng thường có vẻ
rệt ở những BN đã phẫu thuật cắt lách (Xem
ngoài màu nâu nhẹ (light bronze) – hậu quả kết
Hình 12). Ở những BN mang KG đồng hợp tử
hợp của da xanh niêm nhạt của thiếu máu, vàng
βthalassemia là β0/β0, gần như tất cả Hb là HbF.
da do tán huyết trong hệ võng nội mô và xạm da
do ứ sắt mạn tính. Tăng sản TB dòng hồng cầu ồ
ạt (massive erythroid hyperplasia) được mở
Những BN β+/β0 hoặc β+/β+ có một lượng HbA
rộng vào các khoang tủy xương trong hộp sọ gây
thay đổi đi kèm theo HbF tăng. Do cả hai đều
ra biến dạng như mở rộng xương trán và/hoặc
được tăng cường hấp thu sắt từ đường ruột và
xương hàm trên. Sự mở rộng của hàm dưới có
điều trị truyền máu nên sắt huyết thanh, độ bão
thể dẫn đến trật khớp thái dương - hàm. Do liên
hòa transferrin và ferritin đều tăng cao.
quan đến nhiều cơ quan và hệ cơ quan của cơ
thể, các triệu chứng bệnh rất đa dạng và phức tạp
Để chẩn đoán trước sinh bệnh β-thalassemia thể
nhưng thường tập trung vào tình trạng thiếu
đồng hợp tử hoặc dị hợp tử phức hợp, phương
máu và suy tim. Cơ tim phì đại do thiếu máu mạn
pháp được sử dụng là phân tích DNA thu được
gây rối loạn dẫn truyền, nhịp tim chậm trừ khi
từ sinh thiết nhung mao màng đệm. Sự rất không
BN được truyền máu đầy đủ. Nếu tình trạng quá
đồng nhất của các KG β-thalassemia khiến cho
tải sắt không được điều trị, BN sẽ tiến triển bệnh
việc chẩn đoán trước sinh trở nên khó khăn về
cơ tim (cardiomyopathy) do ứ sắt gây suy tim,
mặt kỹ thuật. Hơn nữa, ngay cả khi đã tiến hành
rối loạn nhịp đe dọa đến tính mạng, cùng với xơ
xét nghiệm và chẩn đoán, tính không chắc chắn
gan ứ sắt và suy nội tiết, đặc biệt là tuyến yên và
về mức nghiêm trọng trên lâm sàng, tôn giáo và
tuyến sinh dục. Sự mở rộng của tủy sinh hồng
văn hoá và sự phát triển của những liệu pháp
cầu vào khung xương ngoại vi dẫn đến chứng
mới và tốt hơn có thể ngăn cản cha mẹ không từ
loãng xương, và đôi khi là bệnh lý gãy xương tại
bỏ việc mang thai. Mắc dù vậy khi nghiên cứu
xương dài (Xem Hình 11).
một số khu vực dân cư, chẩn đoán trước sinh đã
có hiệu quả rõ rệt trong việc giảm đáng kể số trẻ
sinh ra mắc bệnh β-thalassemia thể nặng.
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 11
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
Hình 12: Phết máu của một BN mắc bệnh
Hình 13: Biểu đồ sống sót và khỏi hoàn toàn
thalassemia thể nặng trước (A) và sau (B) khi
bệnh (thalassemia-free) của 866 BN mắc bệnh
phẫu thuật cắt lá lách.
βthalassemia thể nặng sau khi được cấy ghép
Phương pháp điều trị
TB gốc từ người hiến giống nhau về HLA (HLAidentical).
Hiện nay, phương pháp điều trị chính của bệnh
Mặc dù liệu pháp truyền máu và thải sắt kéo dài
β-thalassemia là truyền máu và thải sắt. Bên
có hiệu quả rõ rệt trong việc ngăn ngừa các biến
cạnh đó, một số biện pháp khác cũng được áp
dụng trong điều trị thalassemia cho những
chứng bệnh tật như trên, nhưng nó không chỉ trường hợp cụ thể.
gây trở ngại cho BN và người chăm sóc mà còn
đặt ra gánh nặng kinh tế rất lớn. Cấy ghép TB •
Truyền máu – thải sắt: Bệnh nhân mắc
gốc là một giải pháp thay thế khả thi hơn.
thalassemia tùy mức độ sẽ bị thiếu máu mạn tính •
và cần phải truyền máu định kỳ. Đặc biệt, BN mắc
Cấy ghép tế bào gốc: Đây là phương
pháp điều trị hiện đại và có thể chữa khỏi bệnh.
bệnh β-thalassemia thể nặng cần được chăm sóc
đa chuyên ngành đầy đủ. Phương pháp điều trị
Tuy nhiên khả năng có người cho phù hợp để cấy
chính là truyền đủ lượng hồng cầu lắng để duy trì
ghép là rất thấp và chi phí điều trị điều trị của
phương pháp này cũng rất tốn kém. Bên cạnh đó,
nồng độ Hb trong máu trên 10 g/dL. Một liệu
với những bệnh nhân đã bị nhiễm sắt nặng tại các
trình truyền máu phù hợp sẽ ngăn ngừa những
bộ phận như gan, tim… thì tỷ lệ thành công sẽ
biến chứng biến dạng xương, tăng cường sự tăng
trưởng và phát triển của cơ thể, và ngăn ngừa suy
thấp hơn. Biểu đồ sống sót của một số lượng lớn
tim cung lượng cao do thiếu máu nặng. Tuy
BN mắc bệnh β-thalassemia thể nặng ở Ý cho thấy
khoảng 3/4 số BN được cấy ghép thành công mà
nhiên, truyền máu có nguy cơ lây nhiễm HIV và
không để lại hậu quả nghiêm trọng trong khoảng
HBV, HCV,… và nguy cơ tai biến truyền máu như
thời gian lên tới 20 năm. Những BN này có thể
truyền nhầm nhóm máu, phản ứng phản vệ, hình
thành dị kháng thể, quá tải thể tích tuần hoàn….
được coi là khỏi bệnh. Hơn nữa, mặc dù chi phí
Đáng lo ngại hơn nữa khi truyền máu, không thể
tài chính đầu tư ban đầu lớn, cấy ghép TB gốc có
hiệu quả cao về mặt chi phí theo thời gian. Hạn
tránh khỏi việc làm tăng tốc độ quá tải sắt. Do đó,
chế chính là chỉ khoảng 25% BN thalassemia có
BN phải được điều trị bằng thuốc thải sắt đủ
người cho TB gốc tương thích.
lượng để đưa về trạng thái cân bằng sắt âm tính
(negative iron balance) nếu có chỉ định. Mục đích •
Cắt lách: Được chỉ định trong các trường
để chống quá tải sắt nhằm hạn chế biến chứng
hợp: khi bệnh nhân tăng nhu cầu truyền máu (đã
trên các tổ chức, cơ quan trong cơ thể. Bệnh nhân
truyền hồng cầu lắng > 200 -220 ml/kg/năm);
thường phải duy trì dùng thuốc thải sắt cả đời.
Lách to gây cản trở sinh hoạt hàng ngày của
Gần đây, sự phát triển các thuốc thải sắt đường
người bệnh hoặc gây đau; Giảm bạch cầu hoặc
uống hiệu quả có thể làm giảm nhu cầu tiêm dưới
tiểu cầu do cường lách.
da thuốc thải sắt hàng ngày. •
Chăm sóc toàn diện: Là một trong
những ưu tiên hàng đầu với bệnh nhân mắc ꞵ-
thalassemia , việc chăm sóc toàn diện sẽ phòng
ngừa và hạn chế các biến chứng của bệnh, giúp
người bệnh có chất lượng cuộc sống tốt hơn.
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 12
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363 •
Điều trị biến chứng: Tùy theo biểu hiện,
hoặc đồng hợp tử Hb E có TB nhỏ nhược sắc
việc điều trị biến chứng sẽ được điều trị dựa vào
nhưng không có biểu hiện lâm sàng đáng kể
tình trạng bệnh nhân mắc phải như có suy tuyến
nào. Ngược lại, những người mang KG dị hợp
nội tiết, bệnh nhân mắc đái tháo đường, suy tim,
tử phức hợp βE/β0-thalassemia thường có kiểu
xơ gan, loãng xương, rối loạn đông máu…
hình nghiêm trọng không thể phân biệt được
Các phương pháp thay thế để đảo ngược cơ chế
với kiểu hình của β-thalassemia thể nặng. Do
sinh lý bệnh của bệnh β-thalassemia thể nặng bao
tần suất gen của βE rất cao ở vùng Đông Nam Á
gồm phát triển các tác nhân dược lý kích thích cho
đông dân cư, nên βE/β-thalassemia thường
gen γ-globin được biểu hiện và, xa hơn nữa là liệu
xuất hiện và là nguyên nhân chính gây ra bệnh tật và tử vong sớm.
pháp gen hiệu quả. β-thalassemia thể trung gian
Tần số gen của βS tương đối cao ở Trung Phi, Ả
Rập và Ấn Độ. Theo đó, thể hợp tử phức hợp
Một số BN thừa hưởng hai gen β-thalassemia từ β
cả bố và mẹ, song lại có biểu hiện lâm sàng và biến
S/β-thalassemia thường xuất hiện ở những
vùng này cũng như ở những vùng mà người
chứng ít nghiêm trọng hơn so với bệnh
Trung Phi đã di cư đến, chẳng hạn như Ý và Hy
βthalassemia thể nặng và được xác định là mắc
Lạp. Vì alen β-thalassemia cho phép tổng hợp
bệnh β-thalassemia thể trung gian. Những
ít hoặc không tổng hợp β
nguyên nhân giải thích kiểu hình bệnh bao gồm: A, nên thành phần Hb trong TB hồng cầu β gen β
S/β-thalassemia là Hb S, và
+-thalassemia cho phép tổng hợp Hb A; gen
do đó tăng khả năng TB biến thành hình liềm
γ-globin được biểu hiện với tần suất cao; hoặc
lên rất nhiều. BN có KG β
đồng di truyền cùng với bệnh α-thalassemia - S/β0 có biểu hiện lâm
sàng nghiêm trọng như BN mắc bệnh hồng cầu
giúp cải thiện tình trạng mất cân bằng chuỗi hình liềm (β
globin. Theo định nghĩa, BN mắc bệnh
S/βS), trong khi đó những người có KG β
βthalassemia thể trung gian không phụ thuộc vào
S/β+ có biểu hiện nhẹ hơn đáng kể.
truyền máu. Tuy nhiên, họ thường có một số triệu
chứng do thiếu máu và hầu như luôn tiến triển BỆNH α-THALASSEMIA
nặng tình trạng quá tải sắt và biến dạng xương.
Tương tác của β-thalassemia với các biến Tổng quan bệnh
thể cấu trúc Hb phổ biến
đều giữa các gen α-globin liền kề. Bởi vì mỗi
Hai biến thể cấu trúc Hb phổ biến nhất là Hb E
người thừa hưởng một cặp domain gen α-
(α2β226glu→lys) và Hb S (α2β26glu→val). BN mang KG
globin từ bố và một cặp khác từ mẹ (cụm gen
dị hợp tử thừa hưởng gen β-thalassemia từ bố
α-globin chứa 1 gen ζ-globin và 2 gen α-globin
hoặc mẹ và thừa hưởng một trong những biến
trên mỗi nhiễm sắc thể 16) nên có đến bốn
thể cấu trúc trên từ người còn lại; trường hợp
dạng bệnh αthalassemia, tùy thuộc vào số
này thường xuyên xuất hiện và thường có kiểu
lượng gen đã bị mất.
hình lâm sàng nghiêm trọng. Đột biến điểm βE
thay thế bazơ ở vùng ranh giới giữa exon 1 và
intron 1, dẫn đến hạn chế khả năng cắt nối và
do đó làm giảm tổng hợp β-globin. Theo đó, Hb
E tạo ra một kiểu hình thuộc vào dạng rất nhẹ
của β+-thalassemia. Những người dị hợp tử
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 13
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
KG đơn bội (haplotype) bất thường phổ biến
Đông Nam Á nhưng hiếm gặp ở những nơi khác
nhất là –α, trong đó chỉ có một gen α hoạt động. (Xem Hình 15).
Khoảng 30% người da đen châu Phi mang KG
dị hợp tử chứa haplotype này. Do đó, họ bị mất
một trong bốn gen α-globin. Ở phía nam Địa
Trung Hải và Đông Nam Á, haplotype –α cũng
rất phổ biến. Diplotype – – là dạng đột biến mất
cả hai gen α-globin nối tiếp. KG này phổ biến ở
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 14
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
Hình 16: Cơ chế đột biến cấp độ sinh học phân tử
của bệnh α-thalassemia.
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 15 Tình trạng Số Mô tả
Genotype MCV Điện di gene (fL) huyết α- sắc tố globin
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363 thiếu hụt Bình thường 0 αα/αα 80- 2.5% 95 HbA2 Silent carrier 1 -α/αα 72- 2.0% (thể ẩn) 82 HbA2 α- 2 -α/-α hoặc 65- 1.5% thalassemia αα/-- 78 HbA2 trait Bệnh HbH 3 -α/-- 60- 10% β4 72 (HbH) Phù thai 4 --/-- 60- >90% 75 γ4 (Hb Bart)
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 16
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
Bảng: Tổng quan về các loại α -thalassemia.
Các dạng bệnh và triệu chứng lâm sàng
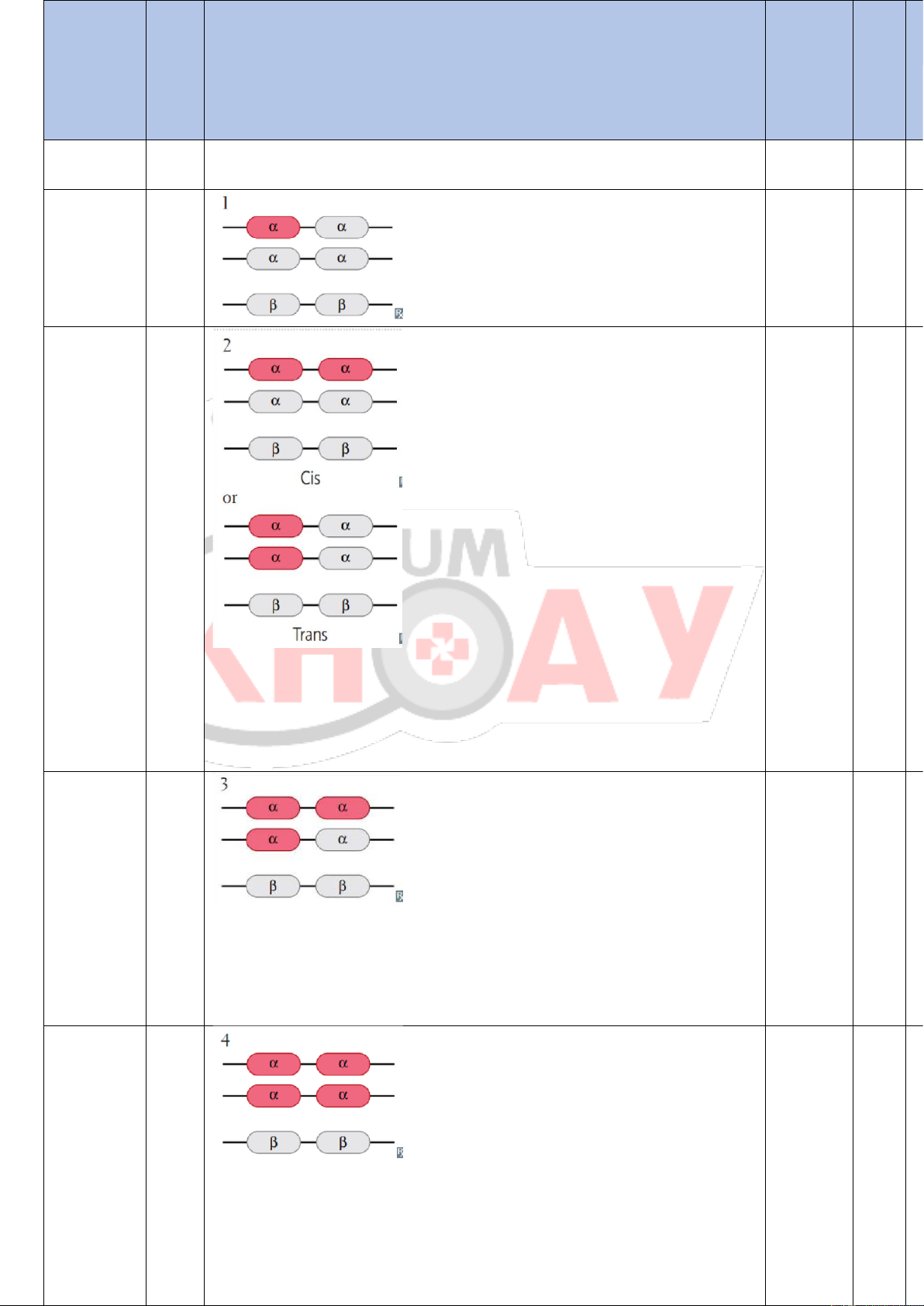
Các KG khác nhau trong bệnh α-thalassemia (–
α/αα, –α/–α, – –/αα, – –/–α, và – –/– –) có mối
tương quan với mức độ biểu hiện kiểu hình trên
lâm sàng dựa trên số lượng gen α-globin bị mất.
Những người có một gen α-globin bị mất (–
α/αα) gọi là “silent carrier”. Về mặt lâm sàng, họ
có kiểu hình hoàn toàn bình thường; ngoại trừ
như trong Hình 5, xét nghiệm số lượng globin
được tổng hợp trong hồng cầu lưới cho thấy tỷ
Hình 17: Phết máu của BN mắc bệnh Hb H – bệnh
lệ β/α tăng nhẹ so với bình thường. Những
α-thalassemia có ba đột biến mất gen (– –/–α).
người mang đột biến mất hai domain gen
αglobin có thể là thể đồng hợp tử chứa hai alen
Do sản xuất chuỗi α-globin giảm mạnh, chuỗi
– α hoặc thể dị hợp tử chứa một alen – –. Hai kiểu
γglobin dư thừa tích tụ trong các TB hồng cầu
bệnh này có kiểu hình lâm sàng giống nhau:
của bào thai (fetus), trẻ mới sinh (newborn) và
hoàn toàn không có triệu chứng, có nồng độ
trẻ sơ sinh (infant); chúng tập hợp lại để tạo
huyết sắc tố bình thường hoặc gần bình thường,
thành một tetramer Hb bất thường được gọi là
hồng cầu nhỏ rõ rệt và tăng tỷ lệ chuỗi β/α.
Hb Bart (γ4). Về sau khi trẻ khoảng 6 tháng tuổi
sau sinh sẽ xảy ra sự chuyển dịch trong biểu
Như được Hình 14, các dạng bệnh αthalassemia
hiện gen globin (từ γ-globin sang β-globin), TB
nghiêm trọng hơn đòi hỏi sự di truyền của một
hồng cầu chứa một lượng lớn β-globin dư thừa;
alen – – kết hợp với một alen – α (bệnh HbH, – –
các chuỗi globin này tự kết hợp lại với nhau để
/–α) hoặc với một alen – – khác (bệnh phù nhau
tạo thành một loại Hb bất thường khác là HbH
thai, hay bệnh Hb Bart/hydrops fetalis, – –/– –)
(β4). HbH có tính hoà tan tương đối kém nên kết
và do đó hầu như bệnh chỉ xuất hiện ở cộng đồng
tủa đáng kể trong các giai đoạn phát triển hồng
người Đông Nam Á. Mặc dù bệnh HbH và Hb Bart
cầu trong tủy xương, gây tán huyết nội tủy và
ít gặp hơn nhiều so với các dạng nhẹ của bệnh α-
tạo thành các thể kết tủa nội bào được gọi là thể
thalassemia, nhận thức về những tình trạng này
Heinz, làm suy yếu khả năng biến dạng của hồng
ở Hoa Kỳ đã được nâng cao do dòng người tị nạn
cầu, thường lắng đọng nhiều trong TB chất của
lớn sau Chiến tranh Việt Nam. Những người mắc
hồng cầu già, dẫn đến làm tăng phá hủy sớm bởi
bệnh HbH có hồng cầu nhỏ đáng kể (Xem Hình
các đại thực bào ở hệ võng nội mô như gan, lách,
17), cùng với sự gia tăng đáng kể tỷ lệ chuỗi β/α.
tủy xương. Do đó, những người mắc bệnh Hb H
dễ xuất hiện những cơn tán huyết nặng khi
nhiễm trùng, sau khi dùng thuốc hay khi sinh.
Việc điều trị cho tình trạng này chủ yếu là mang
tính chất hỗ trợ. Nếu tình trạng quá tải sắt
nghiêm trọng xảy ra, nên điều trị thải sắt.
Sự di truyền của hai alen – – từ cả bố và mẹ dẫn
đến mất hoàn toàn khả năng tổng hợp chuỗi
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 17
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363
αglobin và do đó không thể tạo ra HbF và HbA.
nên được dự đoán trước ở các gia đình châu Á
Kết quả là, gần như toàn bộ Hb của thai nhi là Hb
- nơi một hoặc cả hai bố mẹ và đặc biệt là khi
Bart (γ4). Để Hb vận chuyển oxy một cách hiệu
đã sinh một đứa trẻ trước đó. Chẩn đoán trước
quả, nó phải là một heterotetramer bao gồm hai
sinh đáng tin cậy có thể được thực hiện bằng
cặp tiểu đơn vị khác biệt nhau về cấu trúc (ví dụ:
cách phân tích DNA từ sinh thiết nhung mao
α + γ hoặc α + β). Ngược lại, các homotetramer
màng đệm. Nếu phát hiện sớm – –/– – α-
(homo: đồng hợp) như Hb Bart có ái lực rất cao
thalassemia trong thai kỳ, hầu hết các bậc cha
với oxy; nên mặc dù máu của thai nhi mắc bệnh
mẹ sẽ quyết định chấm dứt thai kỳ. Nếu chẩn
– –/– – thalassemia được cung cấp đầy đủ oxy,
đoán không được thực hiện cho đến hết tam
nhưng Hb Bart không giải phóng oxy đến các
cá thứ hai hoặc thứ ba, bào thai có thể được
mô, và do đó thai nhi bị thiếu hụt nghiêm trọng
cứu bằng cách thay máu vào trong tử cung.
oxy tại mô. Khi đó cơ thể sẽ tăng sản xuất hồng
Sau khi sinh, cấy ghép TB gốc nếu thành công,
cầu tại tủy xương và ngoài tủy, dẫn đến gan lách
có thể chữa khỏi bệnh, nhưng trong số những
to, kéo theo tình trạng thiếu máu hồng cầu nhỏ
người hiến tặng, chỉ tìm thấy được 25% số
nhược sắc nặng nề của thai. Khi mô thiếu oxy
người đủ tương thích để hiến TB.
trầm trọng sẽ xảy ra suy tim cung lượng cao và
hoạt động của Vascular endothelial growth
factor – VEGF . Cuối cùng, gây thoát mạch gian
bào và tình trạng phù toàn thân và phù nhau thai
(Xem Hình 18). Thai sẽ chết non trong bụng mẹ
giữa hoặc cuối thai kỳ hoặc vài giờ - vài ngày sau khi sinh.
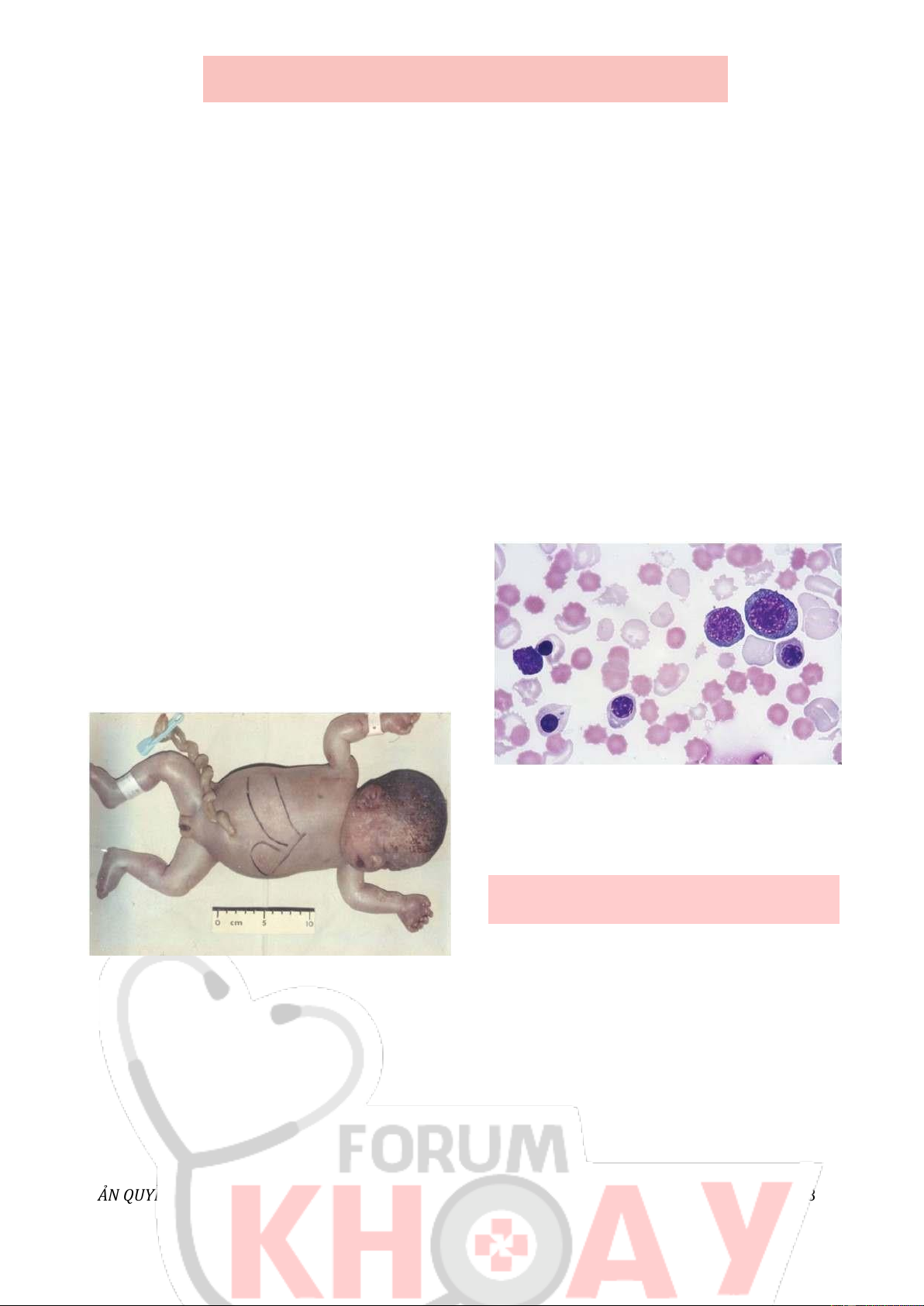
Hình 19: Phết máu của một em bé mới sinh với
tình trạng phù bào thai do bệnh α-thalassemia có
đột biến mất bốn gen (- -/- -). CÂU HỎI ÔN TẬP
Câu 1: Thalessemia là một bệnh di truyền theo kiểu
Hình 18: Hình ảnh em bé chết non với tình trạng
do bệnh α-thalassemia có đột biến mất bốn gen (-
A. Đột biến gen lặn trên NST thường B. -/- -).
Đột biến gen trội trên nst thường
Xem xét hình dạng của hồng cầu cho thấy tình
C. Đột biến gen lặn trên nst giới tính
trạng tăng hồng cầu nhỏ, bất thường về hình D.
dạng hồng cầu và thừa thãi rất nhiều TB hồng
Đột biến gen trội trên nst giời tính
cầu có nhân chưa trưởng thành, (Xem Hình
Câu 2: Chọn liên kết đúng
19). Khả năng gặp phải KG – –/– – ở thai nhi
A. β-thalassemia – NST thứ 16
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 18
CASE PREVIEW: THALASSEMIA lOMoARcPSD| 51038363 B. α-
nhẹ. Xét nghiệ m di truyền cho thấy cô chỉ có
m ột gen bị xoá. Người đàn ông bị thiếu máu
hồng cầu nhỏ và có hai gen bị xoá. Nếu hai D. 75% E. 100% ĐÁP ÁN 1 A 2C 3B 4 C 5C thalassemia – NST thứ 11 B. Tăng HbA2
C. α -thalassemia – Di truyền liên kết gen C. Thiếu máu nặng
D. Thalassemia - Di truyền tính trạng số lượng D. Hồng cầu hình bia
Câu 3: Alpha thalassemia không tương
Câu 5: Một cặp vợ chồng đến trung tâm tư vấn
thích với sự sống vì: A. Hb barts không liên
di truyền trước hôn nhân vì cả hai đều có tiền kết với oxy
sử gia đình mắc bệnh α-thalassemia. Người
phụ nữ có nồng độ hemoglobin giảm
B. Chuỗi β4 tạo thành các hợp chất không hòa tan
C. Oxy bám rất chặt vào Hb barts và không được
giải phóng trong các mô. D. Hồng cầu chặn các mao mạch nhau thai
gen bị xoá của người đàn ông nằm ở hai
nhiễm sắc thể khác nhau (trans), tức một gen
từ mẹ và một gen từ bố, thì khả năng di
truyền hai gen xoá sang con cái của họ là bao nhiêu? A. 0% B. 25% C. 50%
Câu 4: Đặc điểm đúng với Beta-thalassemia trait là A. Tăng HbF
BẢN QUYỀN THUỘC VỀ CLB HỌC THUẬT FORUM KHOA Y 19
Tài liệu liên quan:
-

Câu Hỏi Sinh Di Truyền | Đại học Y Dược Thành phố Hồ Chí Minh
117 59 -

Bài Tập Giữa kỳ Sinh Di Truyền | Đại học Y Dược Thành phố Hồ Chí Minh
83 42 -

Đề thi cuối kì sinh di truyền | Đại học Y Dược Thành phố Hồ Chí Minh
152 76 -

Trắc nghiệm sinh di truyền | Đại học Y Dược Thành phố Hồ Chí Minh
82 41 -

Câu hỏi cuối kì sinh di truyền | Đại học Y Dược Thành phố Hồ Chí Minh
87 44