Cơ Chế Sinh Ung Thư: Khám Phá Sinh Học Phân Tử Ung Thư | Đại học Y Dược Thành phố Hồ Chí Minh
Mô tả các dạng tổn thương DNA và cơ chế sửa chữa. Hiểu được chức năng của tiền gen sinh ung, gen sinh ung và các con đường dẫn truyền tín hiệu tế bào (TB). Hiểu được chức năng của gen đè nén bướu và sự mất dị hợp tử. Hiểu được chức năng của Telomere, Telomerase. Tài liệu giúp bạn tham khảo, ôn tập và đạt kết quả cao. Mời đọc đón xem!
Môn: Phương pháp và kỹ thuật cơ bản trong sinh họcphân tử 11 tài liệu
Trường: Đại học Y Dược Thành phố Hồ Chí Minh 379 tài liệu
Tác giả:





Preview text:
lOMoAR cPSD| 45469857
CƠ CHẾ SINH UNG THƯ
Nguyễn Quốc Bảo, Đoàn Trọng Nghĩa
MỤC TIÊU BÀI GIẢNG
1. Mô tả các dạng tổn thương DNA và cơ chế sửa chữa
2. Hiểu được chức năng của tiền gen sinh ung, gen sinh ung và các con đường
dẫn truyền tín hiệu tế bào (TB)
3. Hiểu được chức năng của gen đè nén bướu và sự mất dị hợp tử
4. Hiểu được chức năng của Telomere, Telomerase
5. Hiểu được cơ chế chết TB theo lập trình
6. Hiểu được cơ chế ngoài gen của sự sinh ung thư 1. MỞ ĐẦU
Ung thư hiện là gánh nặng sức khỏe cho toàn cầu với xuất độ và tử vong ngày
càng tăng nhanh. Theo ghi nhận ung thư toàn cầu năm 2018, ước tính có 18,1 triệu bệnh
nhân mới mắc ung thư và 9,6 triệu người tử vong vì căn bệnh này. Sự sinh ung thư được
xem là một tiến trình đa giai đoạn, phức tạp với sự thay đổi về kiểu hình cũng như ở
mức độ phân tử. Bướu ác tính có một số đặc trưng về mặt kiểu hình như tăng trưởng quá
mức, xâm lấn các mô lân cận và có khả năng di căn xa đến các cơ quan khác. Ở góc độ
phân tử, ung thư là sự rối loạn của các tế bào bên trong cơ thể do sự tích lũy những tổn
thương di truyền, khiến cho những tế bào này có những ưu thế tăng trưởng hơn so với
các tế bào bình thường, hay còn gọi là sự tăng trưởng không kiểm soát được. Trong
phạm vi bài viết này sẽ đề cập đến những cơ chế phân tử của sự sinh ung thư. 2. NỘI DUNG
2.1. Tổn thương DNA và cơ chế sửa chữa
Tổn thương DNA hay còn gọi là đột biến gen là biến cố không thể tránh khỏi
trong quá trình sống. Có nhiều nguyên nhân gây nên các thay đổi về cấu trúc DNA mà
y học ngày càng hiểu rõ hơn. Phần lớn những đột biến gen này là kết quả của sự tiếp xúc
của cơ thể với những tác nhân từ môi trường bên ngoài, được phân thành 3 nhóm: yếu
tố vật lý, hóa học và sinh học. Tia X, tia cực tím là những yếu tố vật lý thường được
nhắc đến và đây cũng là nguyên nhân gây nên nhiều bệnh lý ung thư như ung thư da,
ung thư tuyến giáp… Các yếu tố hóa học thì có rất nhiều, có thể kể đến các hóa chất sinh
ung thư có trong khói thuốc lá (gây ung thư phổi, ung thư hốc miệng, thanh quản…)
hoặc chất Nitrosamine có trong những thực phẩm chế biến dạng muối (muối chua, mắm)
gây ung thư dạy dày. Yếu tố sinh học, cụ thể là virút, vi khuẩn ngoài việc gây nên các 1 lOMoAR cPSD| 45469857
bệnh lý viêm nhiễm, cũng có vai trò trong sự sinh ung thư. Ước tính, virút và vi khuẩn
có liên hệ với khoảng 20% các loại bệnh ung thư, điển hình là virút viêm gan B, C (ung
thư gan), virút sinh u nhú ở người (ung thư cổ tử cung, ống hậu môn) hoặc xoắn khuẩn
môn vị (ung thư dạ dày). Ngoài ra, những tổn thương DNA có thể xảy ra do sự sai sót
trong quá trình nhân đôi tế bào hoặc tế bào tiếp xúc với những sản phẩm hóa học sinh ra
trong quá trình chuyển hóa của cơ thể.
Những tổn thương DNA (đột biến gen) được phân chia thành 2 nhóm lớn là đột
biến về cấu trúc và đột biến về chức năng. Thay đổi về mặt cấu trúc có thể do thêm
nucleotide, mất nucleotide, thay nucleotide hoặc những đột biến phức tạp hơn (pha trộn
nhiều loại đột biến). Tất cả những sự thay đổi này sẽ dẫn đến sự thay đổi về mặt chức
năng của gen, bao gồm: đột biến làm tăng chức năng của gen hoặc làm mất chức năng.
Ước tính, mỗi ngày cơ thể người có thể xảy ra khoảng 104 đến 106 các tổn thương DNA
với những mức độ khác nhau. Nếu như các đột biến gen này được tích lũy dần theo thời
gian sẽ tạo nên những protein có chức năng không phù hợp, gây bệnh lý. Ngoài ra, nếu
đột biến xảy ra ở tế bào mầm sinh dục sẽ có khả năng di truyền cho thế hệ sau.
Nói như vậy không đồng nghĩa với việc xảy ra tổn thương DNA là gây nên bệnh
lý. May mắn thay, cơ thể người đã được trang bị sẵn các cơ chế nhằm phục hồi hoặc loại
bỏ những tổn thương DNA này, mà cơ chế tự sửa chữa DNA là một trong số đó. Mục
đích của quá trình tự sửa chữa DNA của tế bào là giúp đoạn DNA bị tổn thương phục
hồi lại trình tự nucleotide vốn dĩ bình thường trước đây. Có nhiều cơ chế sửa chữa DNA
khác nhau, sẽ được sử dụng tùy thuộc vào những loại tổn thương DNA khác nhau. Có
cơ chế chỉ là đơn giản sửa chữa các bazơ nitơ và cũng có những cơ chế phức tạp hơn
như cắt bỏ đoạn DNA bị tổn thương và dùng nhánh đối diện để tạo nên trình tự DNA
bình thường, dựa theo nguyên tắc bổ sung. Tuy nhiên không phải lúc nào các cơ chế này
cũng hoạt động hoặc hoạt động có hiệu quả. Nếu như điều này xảy ra thì các tổn thương
DNA sẽ không được sửa chữa, lâu dần cơ thể sẽ tích lũy đủ các đột biến gây bệnh. 2 lOMoAR cPSD| 45469857
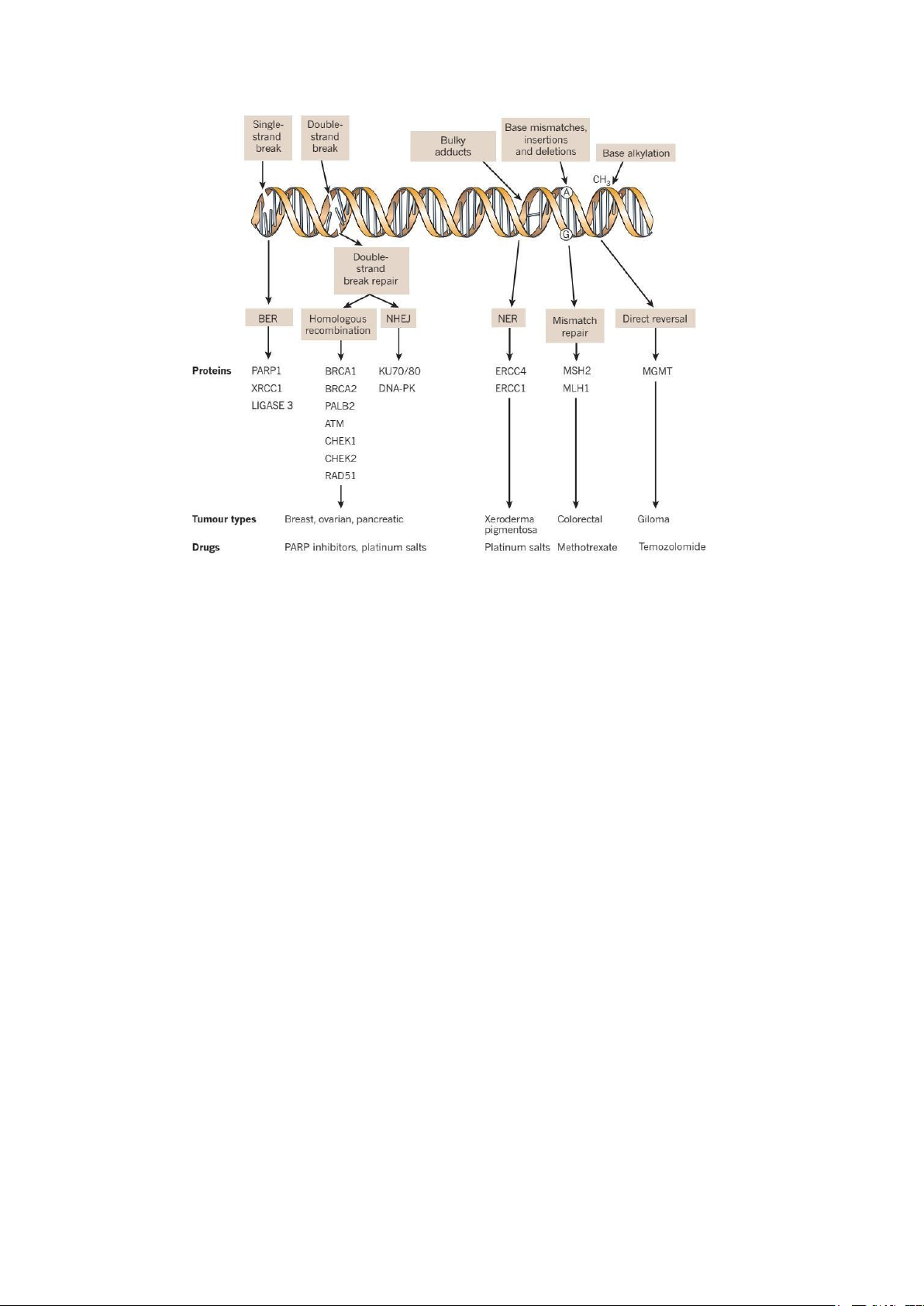
Hình 1: Các loại tổn thương DNA và những cơ chế / protein tham gia sửa chữa
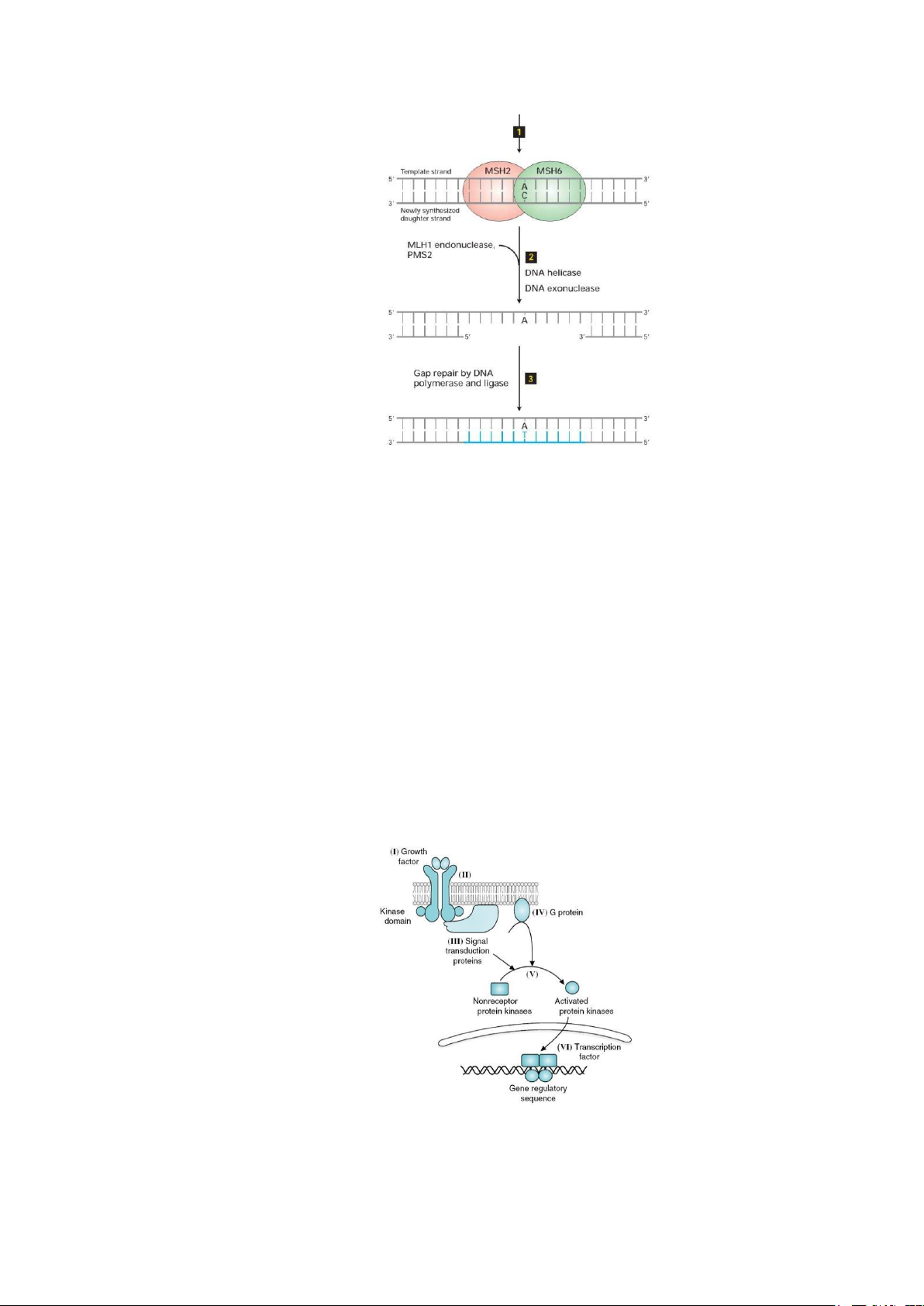
Lấy ví dụ là cơ chế sửa chữa sự bắt cặp sai (MMR: miss-match repair), có thể gặp
trong hội chứng Lynch (hay ung thư đại trực tràng di truyền không polyp). Hội chứng
Lynch chiếm tỉ lệ 2-5% các trường hợp ung thư đại trực tràng, ngoài ra còn có thể gây
một số loại bệnh lý khác như ung thư nội mạc tử cung, dạ dày, ruột non, tụy, tuyến tiền
liệt và da …. Đây là hội chứng di truyền theo gen trội, làm mất chức năng của protein
sửa chữa bắt cặp sai như MSH2, MSH6, MLH1, PMS2. Các protein này đóng vai trò
quan trọng trong việc sửa chữa các sai lệch bắt cặp của DNA trong quá trình nhân đôi.
Bình thường protein MSH6, MSH2 sẽ đi kiểm tra dọc theo chiều dài đoạn DNA, nếu
như phát hiện ra có sự bắt cặp sai các bazơ nitơ như Adenin với Cytosin sẽ thông báo và
huy động những protein MLH1 và PMS2 đến vị trí này, thực hiện tháo xoắn
DNA và cắt bỏ đoạn DNA bị sai sót. Tiếp theo, men DNA polymerase sẽ giúp tổng hợp nên đoạn
DNA mới dựa vào nhánh đối diện theo nguyên tắc bổ sung. Sau đó, ligase sẽ giúp
gắn đoạn DNA mới tổng hợp vào khuyết hổng trước đó. Trong điều kiện các gen MMR
bị đột biến mất chức năng thì cơ chế sửa chữa này sẽ bị bất hoạt và gây nên bệnh lý. 3 lOMoAR cPSD| 45469857
Hình 2: Vai trò sửa chữa DNA của các protein sản phẩm của gen MMR
2.2. Tiền gen sinh ung – gen sinh ung
Tiền gen sinh ung (proto-oncogene) có trong các tế bào bình thường của cơ thể
và có chức năng liên quan đến tăng sinh tế bào. Khi tiền gen sinh ung bị đột biến theo
kiểu tăng hoạt động và trở thành gen sinh ung. Các gen này sẽ mã hóa cho các protein
sinh ung. Protein sinh ung có thể đóng vai trò của bất cứ thành phần nào trong con đường
dẫn truyền tín hiệu tế bào, bao gồm: yếu tố tăng trưởng (EGF, TGF, FGF…), thụ thể yếu
tố tăng trưởng, yếu tố dẫn truyền tín hiệu tế bào, protein G, những thành phần của chu
kỳ tế bào hoặc yếu tố sao chép. Gen sinh ung bị đột biến do nhiều cơ chế như đột biến
làm chuyển vị gen (chuyển tiền gen sinh ung nằm ngay phía sau vùng khởi động), chèn
đoạn gen sinh ung từ virút, biểu hiện quá mức protein sinh ung do sự khuếch đại tiền
gen sinh ung hoặc gen sinh ung hoặc xảy ra đột biến điểm làm thay đổi cấu trúc và chức
năng của tiền gen sinh ung làm gen sản xuất ra protein sinh ung, tham gia vào quá trình
điều hoà và thúc đẩy sự sinh sản tế bào.
Hình 3: Bản chất của các protein sinh ung
Các protein dẫn truyền tín hiệu nằm ở mặt trong màng tế bào, nhận tín hiệu từ sự
hoạt hóa thụ thể yếu tố tăng trưởng ngoài tế bào và dẫn truyền vào trong nhân tế bào. 4 lOMoAR cPSD| 45469857
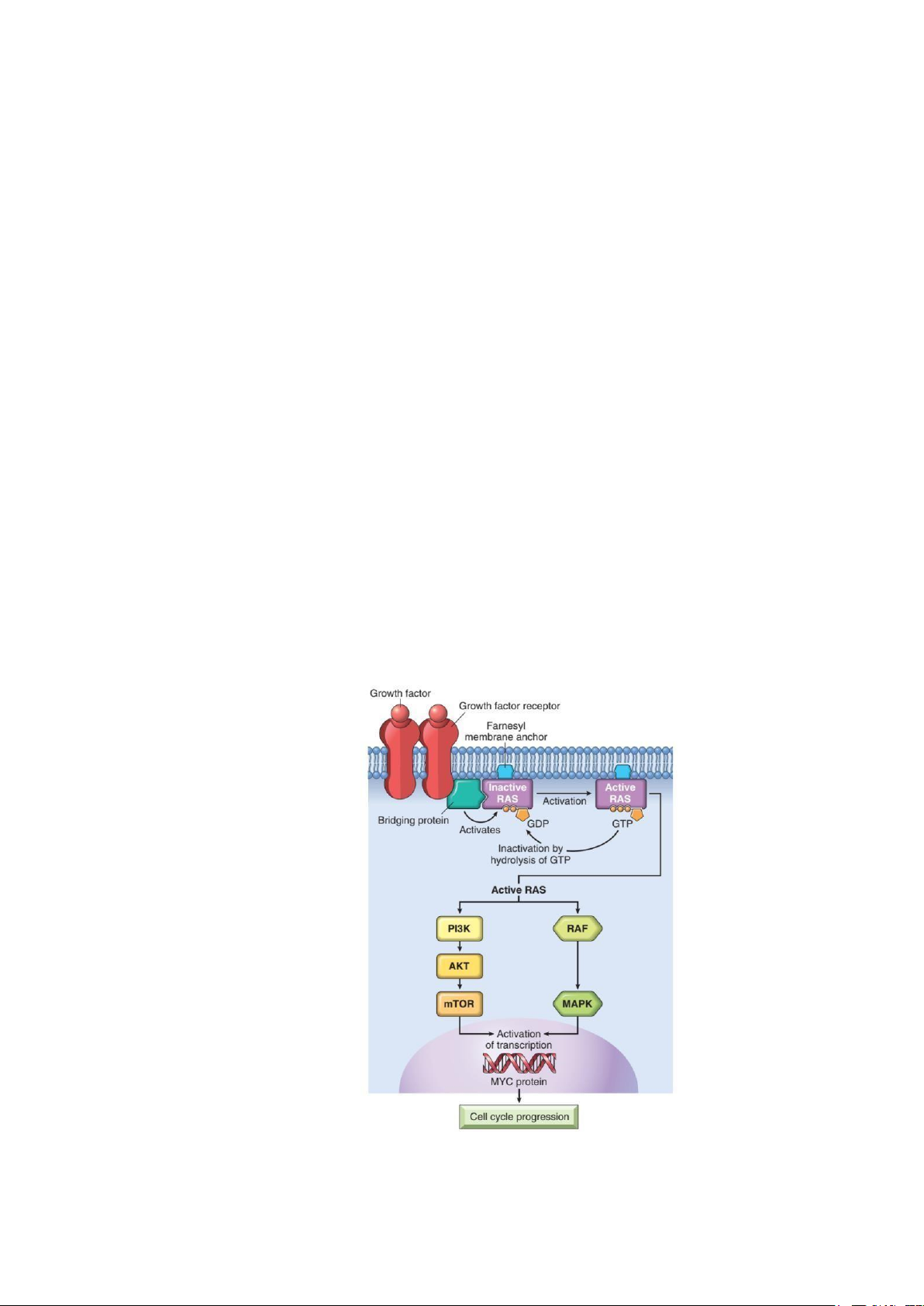
RAS là một protein dẫn truyền tín hiệu và đột biến gen RAS thường gặp trong các bệnh
lý ung thư (15 – 20% trường hợp ung thư ở người). Một số bệnh lý có tỉ lệ cao đột biến
gen RAS như carcinôm tuyến của tụy và carcinôm đường mật (90%), ung thư đại tràng,
nội mạc tử cung và tuyến giáp (50%), carcinôm tuyến ở phổi và bạch cầu dòng tủy
(30%). Dạng đột biến gen RAS thường gặp là đột biến điểm. Khi protein RAS gắn kết
với GDP sẽ ở trạng thái bất hoạt và ngược lại, khi gắn kết với GTP sẽ chuyển thành trạng
thái hoạt hoá. Protein RAS khi được hoạt hóa sẽ hoạt động trên con đường dẫn truyền
MAP kinase, thu hút protein RAF-1. MAP kinase hoạt hóa yếu tố sao chép nhân, thúc
đẩy sự phân bào. Trong tế bào bình thường, sự hoạt hóa protein RAS là thoáng qua do
men GTPase nội sinh sẽ thủy phân GTP thành GDP để chuyển RAS từ trạng thái hoạt
động thành không hoạt động. Vì vậy, mà tín hiệu phân bào là không liên tục. Ở tế bào
mang gen RAS bị đột biến thì protein RAS được hoạt hóa liên tục do mất khả năng thủy
phân GTP nên sẽ kích thích tế bào tăng sinh liên tục. Một ví dụ khác về protein sinh ung
đóng vai trò là thụ thể yếu tố tăng trưởng. Protein RET được mã hóa từ tiền gen sinh ung
RET, đóng vai trò là thụ thể cho tế bào thần kinh đệm, giúp thúc đẩy sự sống còn tế bào
trong quá trình phát triển thần kinh. Bình thường, gen RET biểu hiện ở những tế bào
thần kinh – nội tiết, bao gồm: tế bào cận nang tuyến giáp (tế bào C), tế bào vùng tủy
thượng thận, tiền thân của tế bào tuyến phó giáp. Nếu tiền gen sinh ung RET (chức năng
thúc đẩy sự tăng sinh tế bào) không bị đột biến theo hướng tăng chức năng trong bệnh
lý ung thư mà đột biến theo hướng mất chức năng sẽ gây ra bệnh Hirchsprung (phình
đại tràng bẩm sinh). Ở những bệnh nhân Hirchsprung, đám rối thần kinh ruột không phát
triển được nên sẽ gây ra táo bón kéo dài và tắc ruột.
Hình 4: Cơ chế hoạt động của protein RAS 5 lOMoAR cPSD| 45469857
2.3. Gen đè nén bướu và sự mất dị hợp tử Ngược lại với gen sinh ung, gen đè nén
bướu (tumor suppressor gene) là những gen mã hóa cho những protein có chức năng
kìm hãm sự phân bào. Một số ý kiến cho rằng tên gen đè nén bướu là dùng sai thuật ngữ
do chức năng sinh lý của các gen này là để điều hòa sự tăng trưởng tế bào, chứ không
đơn thuần chỉ là ngăn ngừa sự thành lập bướu. Trong quá trình sinh ung thư, bên cạnh
sự hoạt hoá liên tục các gen có chức năng phân bào (gen sinh ung) thì cũng cần phải bất
hoạt chức năng của những gen có chức năng kìm hãm sự phân bào. Các tác giả đã mượn
hình ảnh của chiếc xe để minh hoạ cho cơ chế hoạt động của gen sinh ung và gen đèn
nén bướu trong quá trình sinh ung thư. Tay ga trên chiếc xe được ví như là gen sinh ung
và tay thắng chính là gen đè nén bướu.
Vậy để khối tế bào tăng trưởng liên tục thì cần tăng cường hoạt động của gen sinh ung
(lên tay ga hết mức) và đồng thời phải bất hoạt chức năng của gen đè nén bướu (gỡ tay
thắng của chiếc xe). Do mỗi gen trong tế bào gồm 2 alen có cùng 1 chức năng nên để
gây mất chức năng gen đè nén bướu cần thiết phải bất hoạt cả 2 alen này. Các cơ chế
làm đột biến mất chức năng của gen đè nén bao gồm: đột biến điểm (tạo ra protein mất
chức năng hoặc sai lệch chức năng), đột biến mất đoạn DNA (làm mất đoạn gen đè nén
bướu và các gen kế cận) hoặc do xảy ra lỗi trong quá trình tách đôi nhiễm sắc thể (dẫn
đến một số tế bào bị mất toàn bộ một nhiễm sắc thể).
Về lịch sử, gen đè nén bướu được phát hiện khi thực hiện nghiên cứu bệnh bướu
nguyên bào võng mạc (bệnh lý hiếm gặp ở trẻ em, tần suất mắc bệnh là 1 / 20.000 trẻ).
Bướu nguyên bào võng mạc xảy ra là do đột biến gen đè nén Rb, nằm trên nhiễm
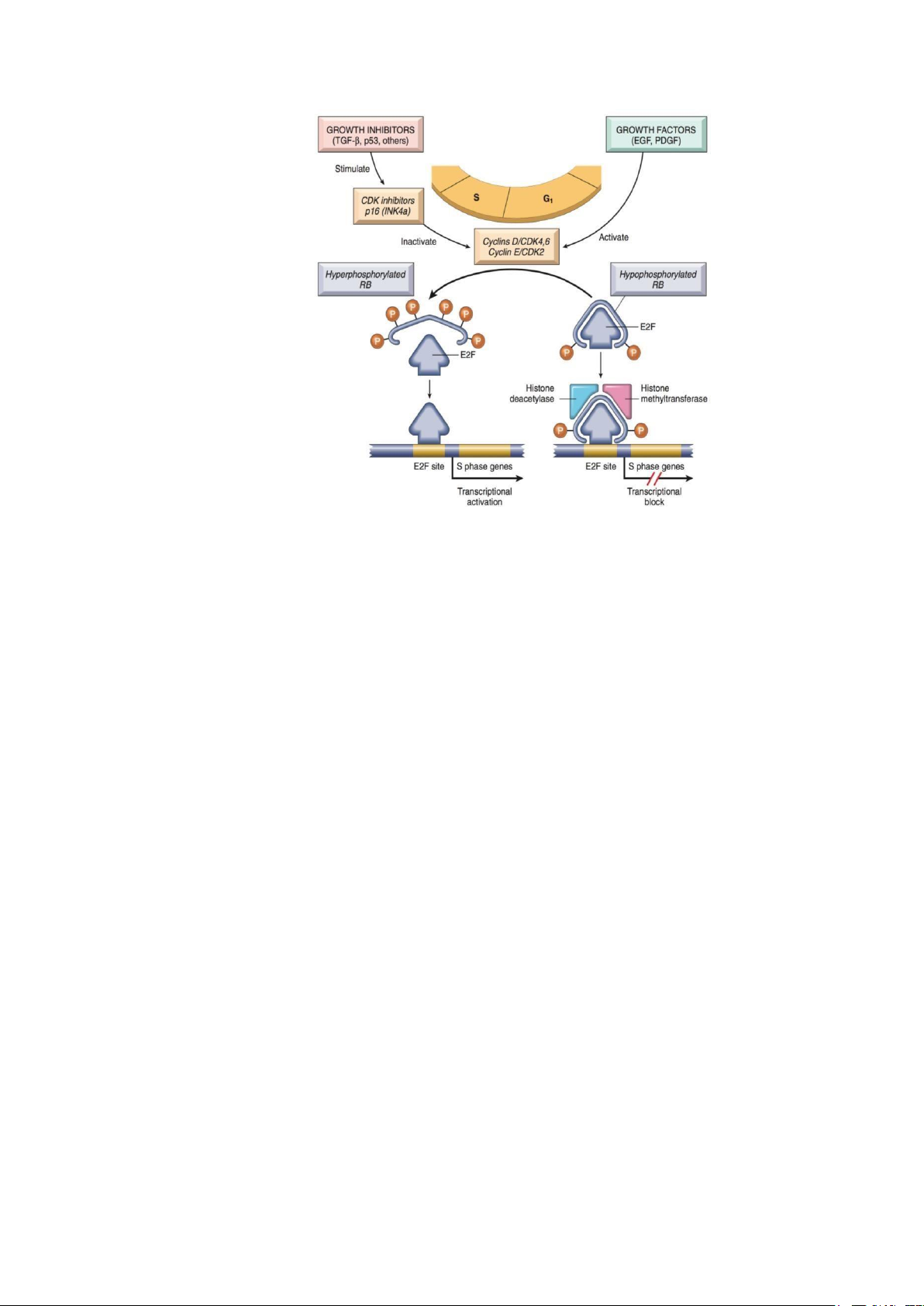
sắc thể 13. Gen Rb mã hoá cho protein Rb, đóng vai trò then chốt trong điều hòa chu kỳ
tế bào. Ở trạng thái hoạt hóa, Rb gắn kết và gây bất hoạt yếu tố sao chép E2F; từ đó sẽ
kìm hãm không cho tế bào từ pha G1 đi vào pha S (điểm kiểm soát G1/S). Ngược lại, ở
trạng thái bất hoạt, protein Rb bị mất chức năng, sẽ làm hoạt hóa yếu tố E2F. Yếu tố E2F
sẽ gắn với vùng khởi động của gen Cyclin E làm tăng biểu hiện protein Cyclin E và sự
tổng hợp phức hợp Cdk2-cyclin E và sau cùng sẽ thúc đẩy tăng sinh tế bào. 6 lOMoAR cPSD| 45469857
Hình 5: Cơ chế hoạt động của gen Rb
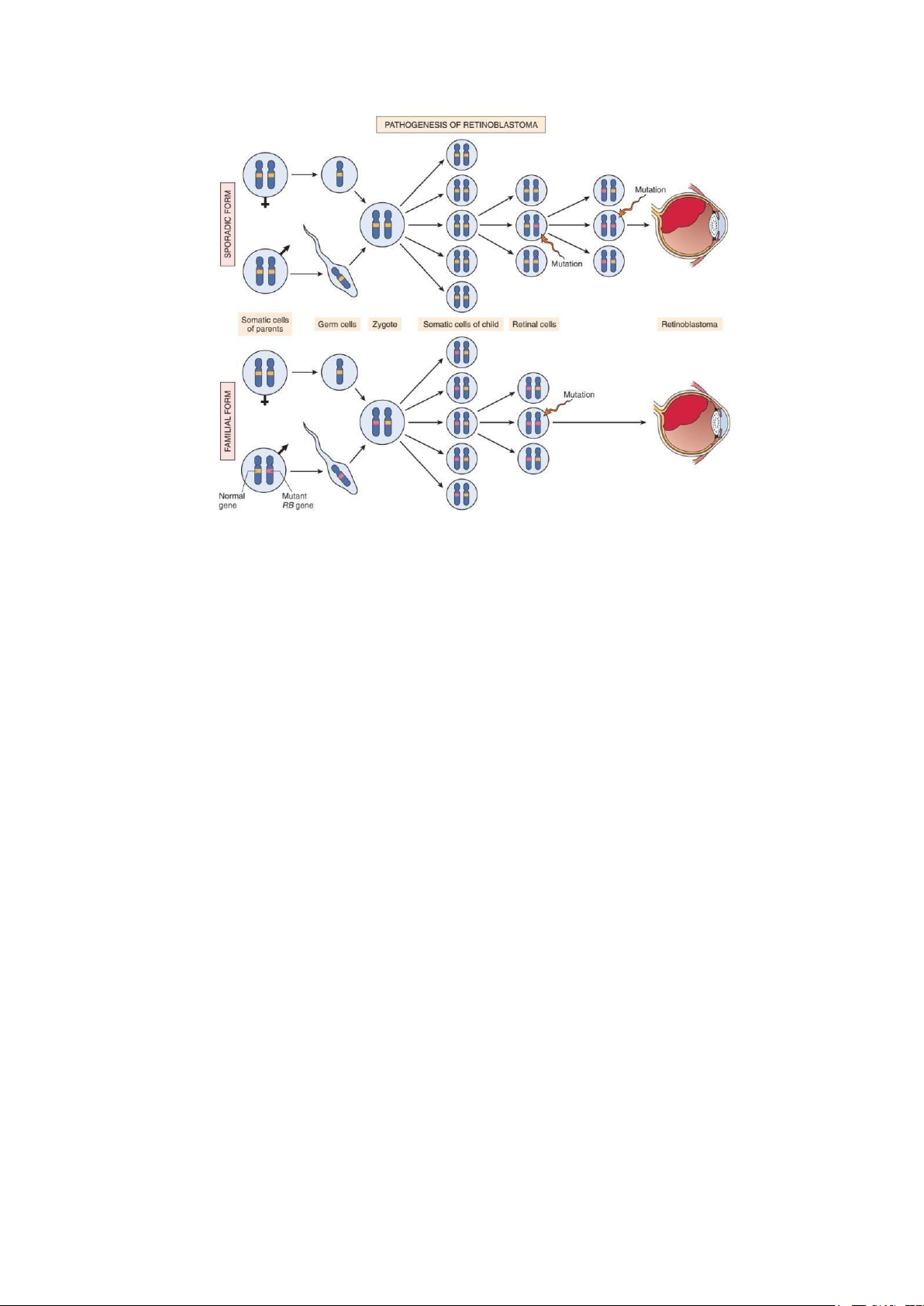
Tác giả Alfred Knudson, năm 1971 đã đề xuất ra giả thuyết “2 cú đánh” để giải
thích cho 2 thể bệnh của bướu nguyên bào võng mạc, gồm thể di truyền (chiếm 1/3 các
trường hợp) và thể đơn lẻ (chiếm 2/3 các trường hợp). Hai cú đánh này là cần thiết và
lần lượt tương ứng cho sự bất hoạt 2 alen của gen Rb. Trong thể bệnh di truyền thì đột
biến đã hiện diện từ giao tử của tế bào cha hoặc mẹ. Khi hợp nhất lại thành hợp tử thì
trong hợp tử này đã mang sẵn 1 alen đột biến. Vì vậy, khi từ tế bào hợp tử này nhân đôi
để tạo ra những thế hệ tế bào sau này thì tất cả các tế bào trong cơ thể khi em bé vừa
chào đời đều có mang 1 đột biến. Đây chính là cú đánh đầu tiên. Vì vậy, trong quá trình
sống sau này theo thời gian, khi tiếp xúc với các yếu tố môi trường, chỉ cần xảy ra thêm
1 đột biến trên bất cứ nguyên bào võng mạc nào cũng có thể tạo nên bướu (cú đánh thứ
2). Trong khi đó ở thể bệnh đơn lẻ thì tế bào hợp tử hoàn toàn bình thường, không mang
đột biến. Do vậy mà tất cả các tế bào được nhân đôi ở thế hệ sau cũng không mang đột
biến alen của gen Rb. Để hình thành nên bướu nguyên bào võng mạc thì cần xảy ra cú
đánh đầu tiên là đột biến gây bất hoạt 1 trong 2 alen của gen Rb ở trong nguyên bào
võng mạc. Tiếp đó, phải cần thêm cú đánh thứ 2 nhằm bất hoạt alen còn lại của gen Rb
của cùng tế bào đó. Do tất cả các thế hệ tế bào trong thể di truyền đều mang sẵn đột biến
1 alen của gen Rb nên thể bệnh này có những đặc điểm thường xảy ra ở trẻ nhũ nhi, tổn
thương thường ở 2 bên mắt và có thể đi kèm với các bệnh lý ung thư khác như sarcôm
xương, sarcôm phần mềm, mêlanôm. Ngược lại, ở thể bệnh đơn lẻ thường xảy ra ở lứa
tuổi muộn hơn (2-5 tuổi), tổn thương thường ở 1 bên mắt và không đi kèm với các bệnh lý ác tính khác. 7 lOMoAR cPSD| 45469857
Hình 6: Bệnh sinh của bướu nguyên bào võng mạc thể di truyền và thể đơn lẻ
Về phân loại, có 2 nhóm gen đè nén bướu gồm nhóm gen gác cổng (gatekeepers)
(Rb / E2F, APC / Beta-catenin) và nhóm chăm sóc bộ gen (caretakers). Nhóm gen có vai
trò chăm sóc bộ gen sẽ tham gia vào việc duy trì tính toàn vẹn của bộ gen thông qua việc
mã hoá những protein thực hiện sửa chữa những tổn thương cấu trúc nhiễm sắc thể,
những thay đổi trong trình tự DNA xảy ra trong quá trình nhân đôi DNA. Trong khi đó
nhóm gen gác cổng mã hoá những protein thực hiện chức năng tại các điểm kiểm soát
trong chu kỳ tế bào nhằm ngăn chặn tế bào bước qua những pha tiếp theo của chu kỳ tế
bào nếu như phát hiện có những bất thường ở tại điểm kiểm soát trước đó hoặc thậm chí
sẽ thúc đẩy tế bào này đi vào con đường chết theo lập trình. Vì vậy sẽ ngăn chặn được
sự tăng sinh tế bào trong những điều kiện bất thường. Gen p53 là gen đè nén bướu được
biết đến nhiều nhất, có vai trò trong hơn 50% trường hợp bệnh ung thư. Đây là gen đa
chức năng, được ví như “người canh gác bộ gen”. Chức năng bình thường của gen p53
bao gồm dừng chu kỳ tế bào, tham gia sửa chữa DNA và thúc đẩy chết tế bào theo lập
trình đối với những tổn thương DNA không thể sửa chữa.
2.4. Telomere và telomerase
Có 2 vấn đề về cấu trúc của nhiễm sắc thể đã được nêu ra trong các bài báo khoa
học. Đầu tiên, các đầu tận tự do của các nhiễm sắc thể có thể kết hợp với các nhiễm sắc
thể kế cận để tạo nên tổ hợp nhiễm sắc thể mới, và đây có thể là một dạng đột biến gen.
Ngoài ra, mỗi khi tế bào thực hiện côn việc nhân đôi thì luôn luôn có một phần nhỏ tại
đầu tận của nhiễm sắc thể không được sao chép và bị mất đi. Vấn đề được đặt ra là nếu
như đoạn nhiễm sắc thể bị mất đi này chứa đựng thông tin di truyền thì điều gì sẽ xảy ra
? Những vấn đề này của nhiễm sắc thể người đã được tạo hóa giải quyết bằng một cấu 8 lOMoAR cPSD| 45469857
trúc, gọi là Telomere. Telomere chính là phần tận cùng của nhiễm sắc thể, gồm những
trình tự giàu Guanin, được lặp đi lặp lại (TTAGGG). Trình tự TTAGGG không mã hóa
cho bất cứ loại protein nào, vì vậy dù nhiễm sắc thể có bị “bào mòn” dần qua các chu
kỳ nhân đôi tế bào cũng không làm mất thông tin di truyền của tế bào. Hơn nữa, tại đầu
tận của nhiễm sắc thể có sự hình thành nên phức hệ protein – telomere (telomere
capping) làm đầu tận cùng nhiễm sắc thể cuộn lại, che chắn đầu tận của nhiễm sắc thể
(như hình ảnh của đầu dây cột giày) khiến cho các nhiễm sắc thể kế cận không thể tự kết hợp với nhau.
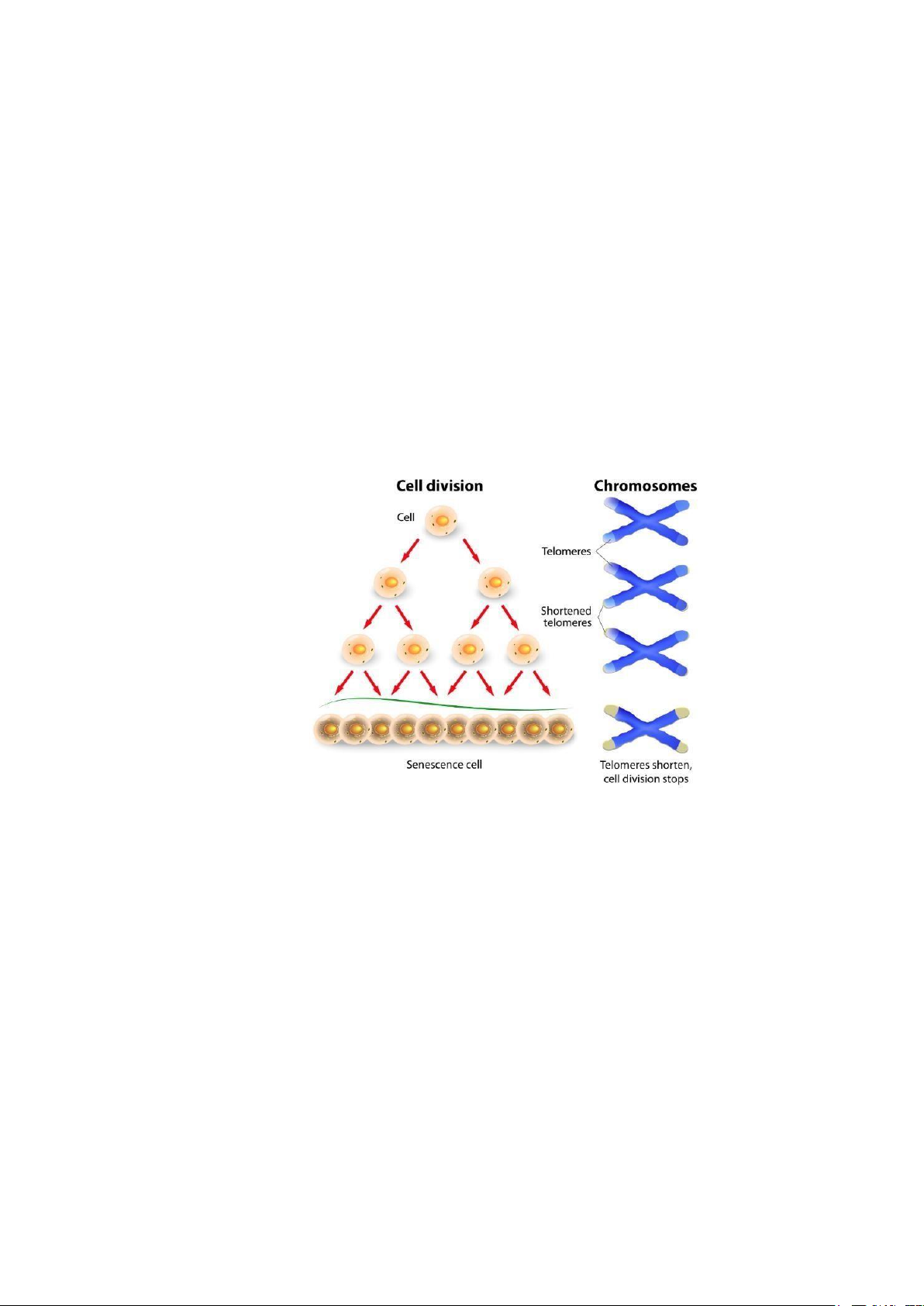
Sinh lý tế bào cho thấy, tế bào trải qua nhiều lần nhân đôi thì đoạn Telomere bị
“bào mòn” dần. Khi đoạn telomere bị “cắt cụt” thì tế bào đó sẽ không thể tiếp tục sinh
sản, nhân đôi được nữa, và sẽ được thúc đẩy vào con đường chết theo lập trình nhằm
bảo vệ sự ổn định thông tin di truyền. Do đó, số lần phân bào của mỗi tế bào là có giới
hạn (giới hạn Hayflick).
Hình 7: Vai trò của telomere trong kiểm soát sự phân bào của tế bào bình thường
Tế bào ung thư dù cho có đặc tính tăng sinh không kiểm soát được, thì lẽ ra số
lần phân bào cũng phải có giới hạn, hay nói cách khác, sự tăng trưởng cũng phải có điểm
dừng khi đoạn Telomere của tế bào ung thư cũng bị “cắt cụt” hoàn toàn. Vậy điều gì đã
khiến cho tế bào ung thư có thêm một đặc tính khác nữa, chính là số lần phân bào là vô
hạn, giúp cho tế bào ung thư có thể tăng sin không ngừng nghỉ. Câu trả lời chính là nhờ
vào một loại men, mang tên là Telomerase. Telomerase là men sao chép ngược (bản chất
là ribonucleprotein), giúp gắn kết trình tự “TTAGGG” vào đầu tận của chuỗi DNA (vùng
Telomere) bằng cách sử dụng chuỗi RNA đơn làm mẫu. Nói cách khác, men Telomerase
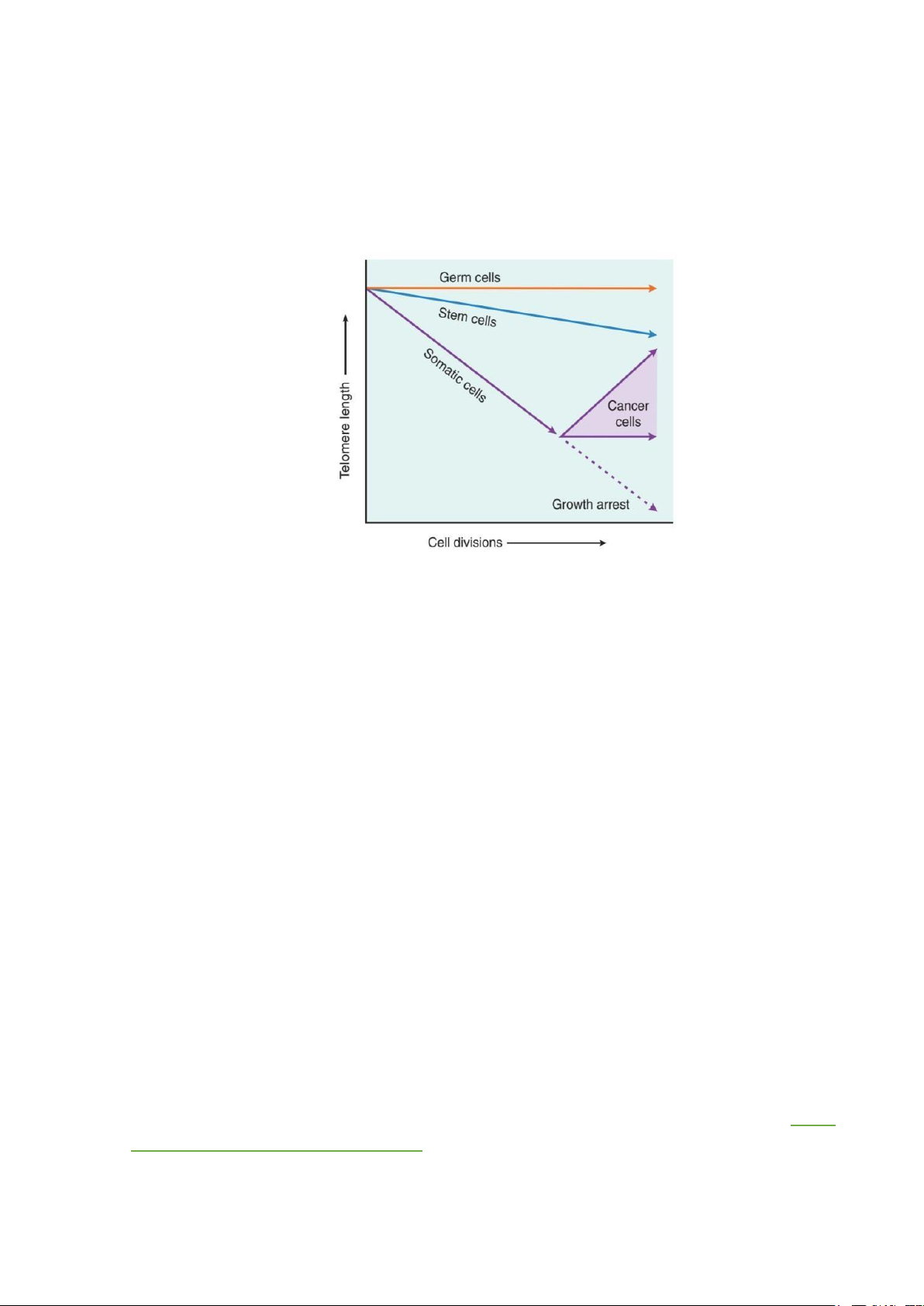
giúp làm tăng chiều dài của đoạn Telomere. Nghiên cứu cho thấy hoạt động của
Telomerase sẽ khác nhau tùy loại tế bào. Ở tế bào mầm (trứng, tinh trùng) và tế bào gốc
đều có telomerase hoạt động nhưng chỉ có tế bào mầm là có đủ nồng độ Telomerase để
duy trì độ dài Telomere. Vì nếu độ dài Telomere của tế bào mầm bị thay đổi thì giống
loài sẽ bị tuyệt chủng. Ở các tế bào sinh dưỡng bình thường thì không có sự hoạt động 9 lOMoAR cPSD| 45469857
của Telomerase, Telomere ngắn dần theo số lần phân bào cho đến khi ngừng tăng trưởng, lão hóa và chết.
Ngược lại, ở tế bào ung thư, Telomerase thường được tái hoạt hóa và hoạt động
mạnh nhất nên chiều dài Telomere luôn được giữ nguyên vẹn như ban đầu. Điều này
làm cho số lần phân bào của những tế bào ung thư là vô hạn.
Hình 8: Hoạt động của Telomerase ở các loại tế bào
Ngày nay, y học cũng đang ứng dụng những hiểu biết về Telomere và Telomerase
vào nghiên cứu điều trị ung thư. Về mặt lý thuyết, có thể sử dụng chất ức chế men
Telomerase ngăn ngừa sự phục hồi lại đoạn Telomere, giúp giới hạn số lần nhân đôi của
tế bào ung thư và khiến các tế bào ung thư sẽ chết như những tế bào khác trong cơ thể.
Ở góc nhìn ngược lại, cũng có thể sử dụng các chất làm hoạt hóa men Telomerase, với
đích nhắm là các tế bào của hệ miễn dịch, để có thể giúp kéo dài đoạn Telomere của
những tế bào này, góp phần chống chọi lại TB ung thư.
2.5. Chết tế bào theo lập trình
Chết tế bào theo lập trình hay còn gọi là Apoptosis (gốc tiếng Hy Lạp có nghĩa là
lá vàng rụng trong mùa thu) là quá trình chết tự nhiên của các tế bào trong cơ thể, được
tác giả Kerr mô tả lần đầu tiên hiện tượng này. Chết tế bào theo lập trình thực chất là
một quá trình điều hòa của cơ thể, cho phép các tế bào tự chết đi nhằm loại bỏ các tế bào
không mong muốn hoặc các tế bào bị mất chức năng. Hiện tượng này có thể diễn ra
trong tự nhiên như quá trình loại bỏ các tế bào ở vùng đuôi nòng nọc khi biến hoá thành
ếch. Ở người trưởng thành, ước tính mỗi ngày có khoảng 50-70 triệu tế bào trải qua chết
theo lập trình. So với kiểu chết tế bào do hoại tử với đặc điểm tế bào bị phồng lên, kèm
sự thoát men tiêu thể, màng tế bào bị vỡ ra và những mảnh vụn sẽ bị tiêu huỷ bởi đại
bào và bạch cầu đa nhân trung tính thì ngược lại trong kiểu chết tế bào theo lập trình, tế
bào sẽ bị co lại, bào tương và màng tế bào bị phân thành từng mảnh nhỏ, nhân tế bào bị
chia nhỏ, chất nhiễm sắc bị cô đặc và có sự thành lập của thể chết theo lập trình. Bảng
1: So sánh đặc điểm giữa chết tế bào theo lập trình và hoạt tử tế bào 10 lOMoAR cPSD| 45469857
Chết tế bào theo lập trình
Hoại tử tế bào
Có vai trò của ty thể và Cytochrome C Không có vai trò của ty thể
Không có sự thoát men tiêu thể
Có sự thoát men tiêu thể
Đặc trưng bởi những biến đổi nhân TB Nhân TB bị mất đi
Thành lập “thể chết theo lập trình” Không có DNA bị chẻ nhỏ Không có
Có sự hoạt hóa men tiêu đạm đặc hiệu Không có
Tiến trình có thể điều hòa
Tiến trình không thể điều hòa
TB chết bị tiêu hủy bởi những TB xung TB chết bị tiêu hủy bởi đại bào và quanh
bạch cầu đa nhân trung tính
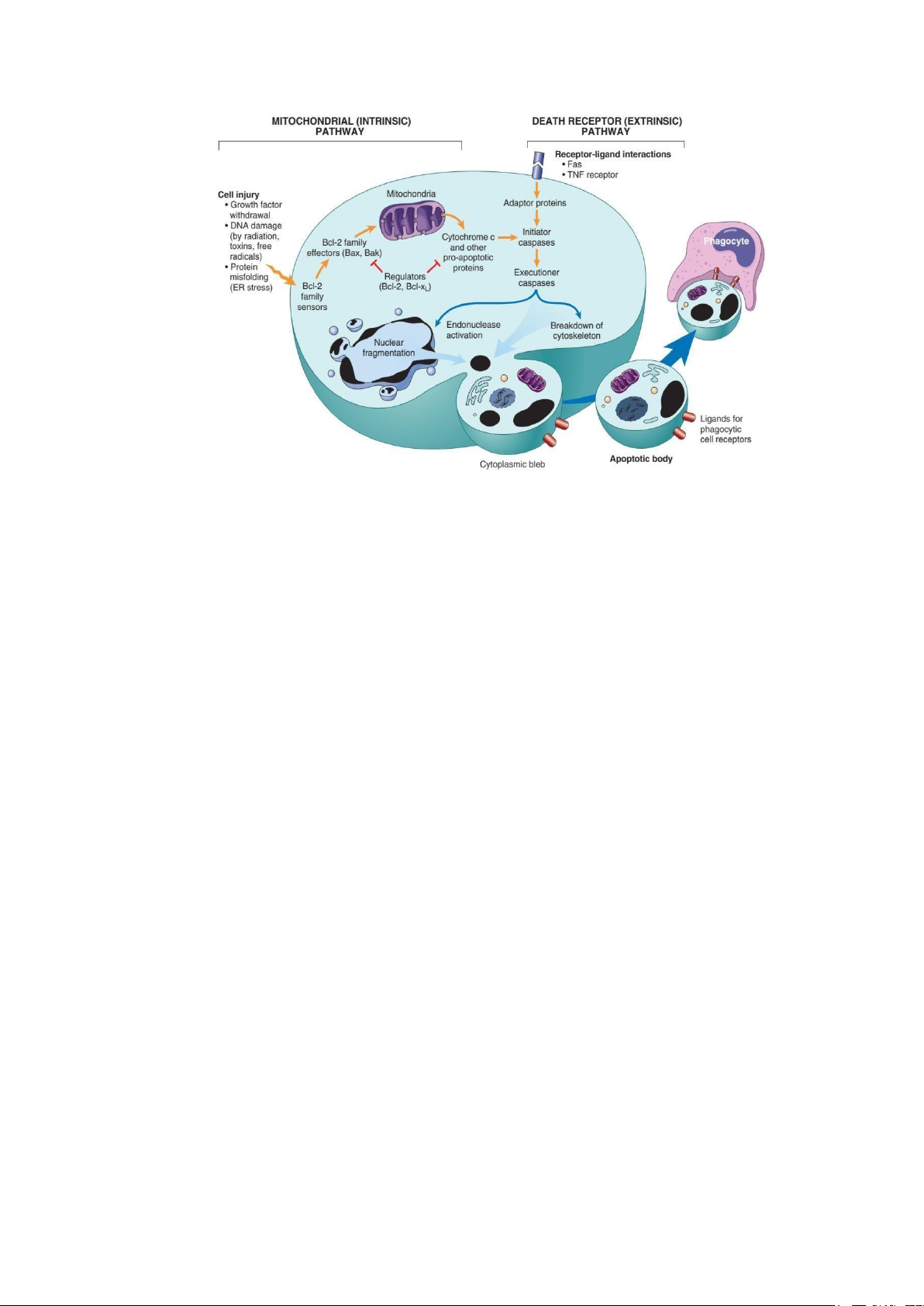
Trong cơ chế chết tế bào theo lập trình thì Caspases là yếu tố rất quan trọng, vừa
đóng vai trò là yếu tố khơi mào và vừa là yếu tố gây chết trực tiếp. Có 2 con đường chính
để kích hoạt Caspase, gồm: con đường nội sinh (thông qua sự hoạt động của ty thể), con
đường ngoại sinh (thông qua hoạt động của các thụ thể gây chết tế bào). Chết tế bào theo
lập trình theo con đường nội sinh được gây ra bởi các kích thích nội bào như các tổn
thương gen không sửa chữa được (do bức xạ, độc tố, các gốc tự do), tình trạng thiếu
Oxy, những stress oxy hóa nghiêm trọng... Con đường chết tế bào theo lập trình nội sinh
này được điều hòa chặt chẽ bởi những protein thuộc gia đình Bcl. Có 2 nhóm protein
Bcl chính là protein tiền chết TB theo lập trình (Bax, Bak, Bad, Bcl-Xs, Bid, Bik, Bim
và Hr) và protein chống lại chết TB theo lập trình (Bcl-2, Bcl-XL,Bcl-W,Bfl-
1 và Mcl-1). Sự điều hoà qua lại của 2 nhóm protein này có thể khởi phát việc tăng tính
thấm của ty thể và phóng thích ra các phân tử tiền phân bào (Cytochrome-C) vào bào
tương nhằm hoạt hoá Caspases. Trong khi đó, để khởi đầu con đường chết tế bào theo
lập trình ngoại sinh cần phải có sự tương tác giữa các phối tử với thụ thể gây chết (Fas
và thụ thể yếu tố hoại tử bướu). Sau đó, bộ đôi này sẽ gắn kết với các protein tiếp hợp
để tạo nên phức hợp tín hiệu gây chết (DISC: Deathinducing signaling complex) để từ
đó hoạt hoá men pro-caspase 8 thành caspase 8. Chính yếu tố caspase 8 sẽ khởi đầu cho
quá trình chết tế bào theo lập trình bằng cách chia ra những Caspase xuôi dòng khác và
những Caspase gây chết trực tiếp. 11 lOMoAR cPSD| 45469857
Hình 9: Cơ chế gây chết tế bào theo lập trình
Bên cạnh đặc tính tăng trưởng tế bào không kiểm soát được, số lần phân bào vô
hạn thì sự mất điều hoà chết tế bào theo lập trình cũng góp phần vào sự sinh ung thư. Sự
mất điều hoà chết tế bào theo lập trình có thể do sự suy yếu con đường dẫn truyền tín
hiệu (giảm biểu hiện thụ thể gây chết, giảm biểu hiện tín hiệu gây chết hoặc biểu hiện
thụ thể “mồi” không có miền gây chết) hoặc do sự mất cân bằng của những protein thuộc
gia đình Bcl (giảm biểu hiện những protein tiền chết theo lập trình hoặc tăng biểu hiện
của những protein chống lại chết theo lập trình). Trực tiếp hơn, sự mất điều hoà chết
theo lập trình này còn do sự giảm biểu hiện của Caspase hoặc do sự đột biến của gen
p53. Như đã đề cập ở phần gen đè nén bướu (mục II.C), gen p53 là gen thường bị đột
biến nhất được tìm thấy trong nhiều loại bệnh lý ung thư ở người. Gen p53 được xem
như là “người canh gác bộ gen” vì đảm nhiệm nhiều chức năng trong cơ thể và một
trong số vai trò của gen p53 là hoạt động mã hóa cho protein Bax (có chức năng bình
thường là thúc đẩy chết tế bào theo lập trình). Vì vậy, những đột biến làm mất chức năng
gen p53 sẽ làm mất biểu hiện protein Bax, khiến cho tế bào không đi con đường chết tế
bào theo lập trình. Một ví dụ về bệnh lý ung thư liên quan đến gen trong cơ chế chết tế
bào theo lập trình là bệnh bạch cầu lymphô mạn. Bệnh lý này có mang đột biến chuyển
vị nhiễm sắc thể [t(14: 18)] làm hoạt hóa gen Bcl-2. Gen Bcl-2 mã hoá cho protein Bcl-
2, bản chất giúp chống lại sự chết tế bào theo lập trình. Do vậy mà trong bệnh bạch cầu
lymphô mạn, tế bào ác tính tuy tăng sinh với tốc độ bình thường nhưng vì những tế bào
này không được chết đi nên số lượng tế bào tích lũy chậm dần theo thời gian. Điều này
hoàn toàn phù hợp với diễn tiến lâm sàng chậm, mạn tính của bệnh.
2.6. Cơ chế ngoài gen của sự sinh ung thư
Như chúng ta đã biết trong tế bào có ít nhất 2 dạng thông tin, gồm những thông
tin về gen để cung cấp các vật liệu tổng hợp protein cho tế bào hoạt động chức năng và 12 lOMoAR cPSD| 45469857
những thông tin ngoài gen có vai trò điều phối, hướng dẫn gen nên được biểu hiện, sử
dụng ở đâu, khi nào và như thế nào ? Năm 1943, tác giả C.H.Waddington đưa ra khái
niệm về epigenetics (ngoài gen). Epigenetics được định nghĩa là ngành học nghiên cứu
những biến đổi biểu hiện gen mà không có sự thay đổi trình tự của chuỗi DNA trong bộ
gen. Bản chất của sự biến đổi biểu hiện gen này là do thông qua các phản ứng hoá hoc
trên chuỗi DNA xảy ra trong quá trình điều tiết sự phiên mã như methyl hóa DNA (DNA
methylation), biến đổi phân tử Histone (Histone modification) và micro RNA (miRNA).
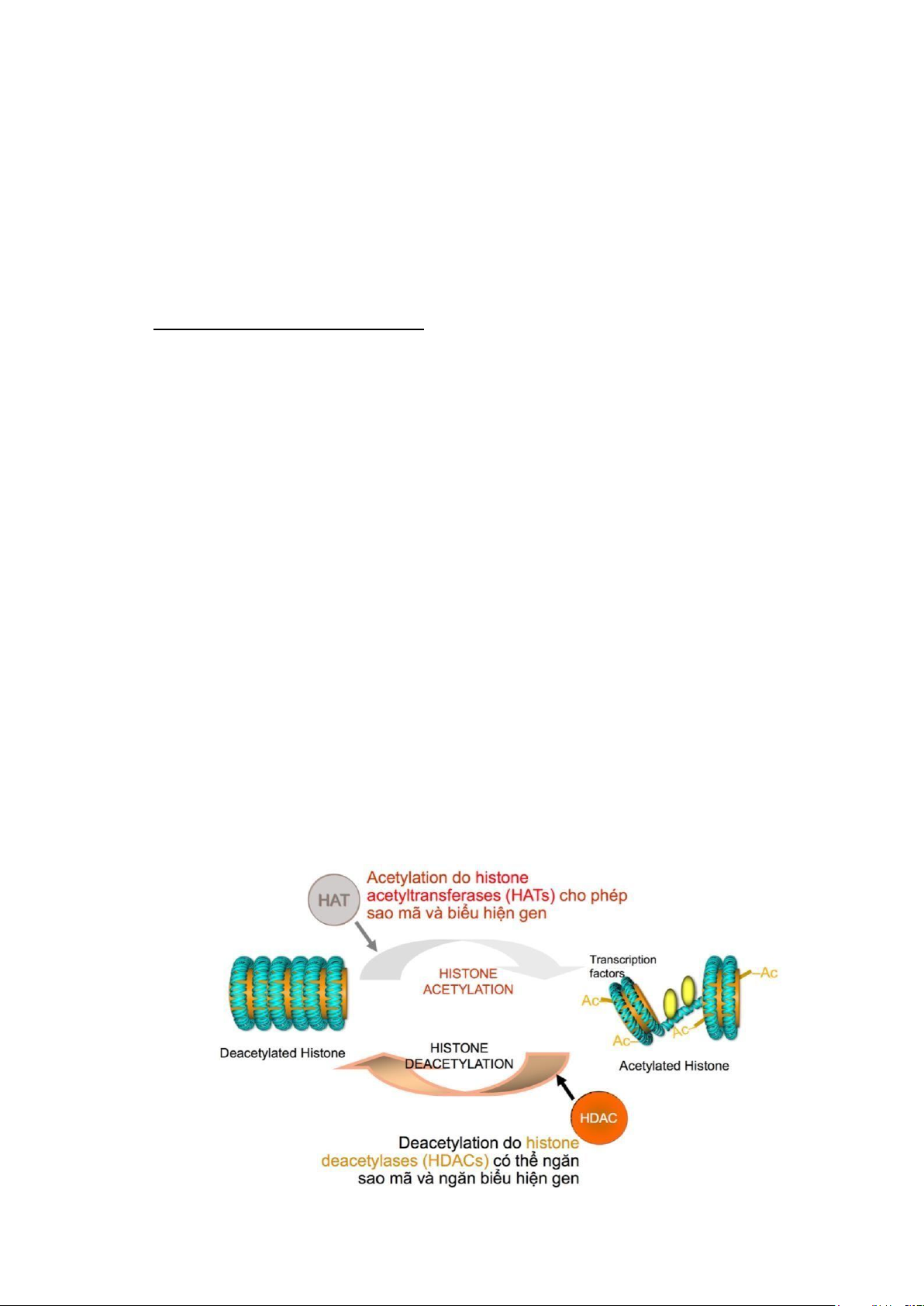
❖ Cơ chế biến đổi phân tử Histone
Trong tế bào, Histone là các protein có chức năng đóng gói và sắp xếp DNA thành
các đơn vị cấu trúc là nucleosome, tạo thành nhiễm sắc chất (chromatin) và nhiễm sắc
thể (chromosome). Vì sợi DNA khi dãn ra sẽ có chiều dài ước tính khoảng 1,8 mét nên
để có thể chứa trong nhân tế bào có kích thước nhỏ thì DNA phải cuộn quanh các thể
Histone, giống như sợi chỉ quấn quanh cuộn chỉ nhằm nén lại kích thước. Chiều dài
DNA sau khi cuộn quanh Histone còn khoảng 0,09 mm, nghĩa là đã giảm đi 20.000 lần.
Đây được xem là dạng đóng của nhiễm sắc thể, với tính chất đông đặc và có rất ít hoạt
động xảy ra trên chuỗi DNA, gen ít được biểu hiện. Tuy nhiên, khi cơ thể cần các protein
hoạt động chức năng thì nhiễm sắc thể sẽ được tháo xoắn để trở thành dạng mở (dạng
hoạt động), giúp gen được biểu hiện để thực hiện việc phiên mã thành mRNA và dịch
mã thành protein. Sự tháo xoắn nhiễm sắc thể hay chuyển nhiễm sắc thể từ dạng đóng
sang dạng mở là nhờ quá trình biến đổi phân tử Histone gồm sự acetyl hoá Lysine,
methyl hoá Lysine và Arginine, phosphoryl hoá serine và threonine... Quá trình biến đổi
này xảy ra tại vị trí đầu N của phân tử Histone và do một số men tham gia. Do vậy những
men này được ví như có vai trò điều hành nhiễm sắc thể. Lấy ví dụ như nhóm men HATs
(Histone acetyltransferases) thực hiện gắn gốc Acetyl (-COCH3) vào vị trí NH3+ của
Lysine giúp loại bỏ điện tích dương của NH3+. Do đó, ái lực giữa Histone và DNA sẽ
giảm đi nên nhiễm sắc thể được tách rời ra, các gen được biểu hiện và thực hiện quá
trình phiên mã. Ngược lại, quá trình khử Acetyl làm nhiễm sắc thể cuộn chặt lại và gen
không được biểu hiện. 13 lOMoAR cPSD| 45469857
Hình 10: Sự biến đổi phân tử Histone
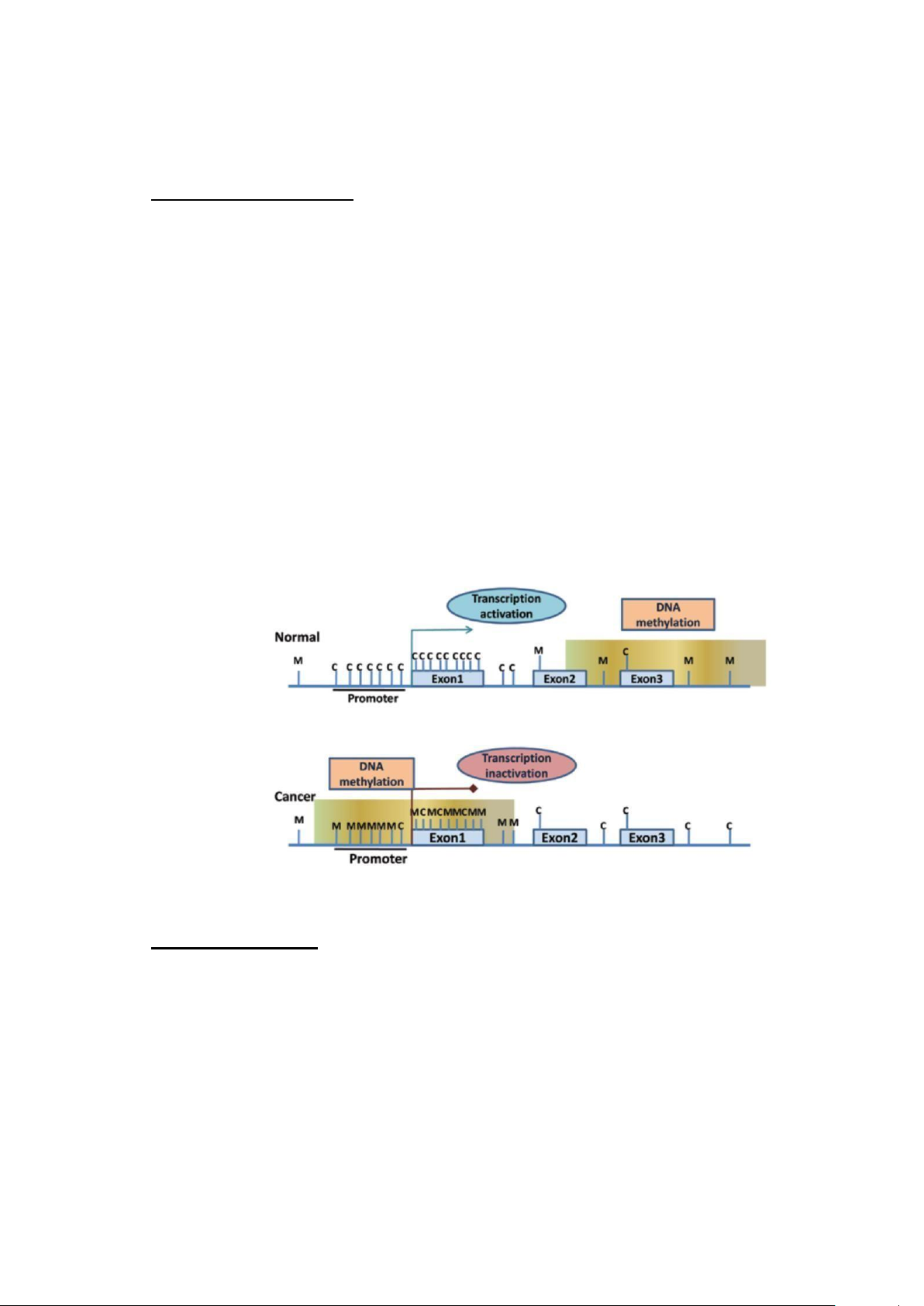
Cơ chế methyl hóa DNA
Sự methyl hóa DNA là cơ chế ngoài gen đơn giản nhất, bằng cách gắn thêm gốc
methyl (-CH3) hoặc acetyl (-COCH3) ở vị trí phân tử Carbon C5 của Cytosine trên chuỗi
DNA. Sự thay đổi này không làm thay đổi trình tự của DNA nhưng lại ngăn không
cho men RNA polymerase II tiếp cận với chuỗi DNA nên không có sự thành lập mRNA.
Ngoài ra, sự methyl hóa DNA tại vị trí CpG còn giúp huy động men HDAC (Histone
deacetylase). Như đã đề cập ờ phần trên thì sự khử Acetyl làm nhiễm sắc thể cuộn chặt
lại và gen không được biểu hiện. Tóm lại, sự methyl hóa DNA cuối cùng sẽ ức chế quá
trình phiên mã, không thể thành lập được mRNA, và do vậy không tổng hợp được các
protein hoạt động chức năng. Phản ứng này được xúc tác bởi những men trong gia đình
DNMT như DNMT1, DNMT3A, DNMT3B. Hiện nay, nhiều loại men điều hành nhiễm
sắc thể được vào thử nghiệm lâm sàng trong nghiên cứu điều trị ung thư như DNMT và HDAC.
Hình 11: Cơ chế methyl hoá tại vị trí Cytosine trên chuỗi DNA
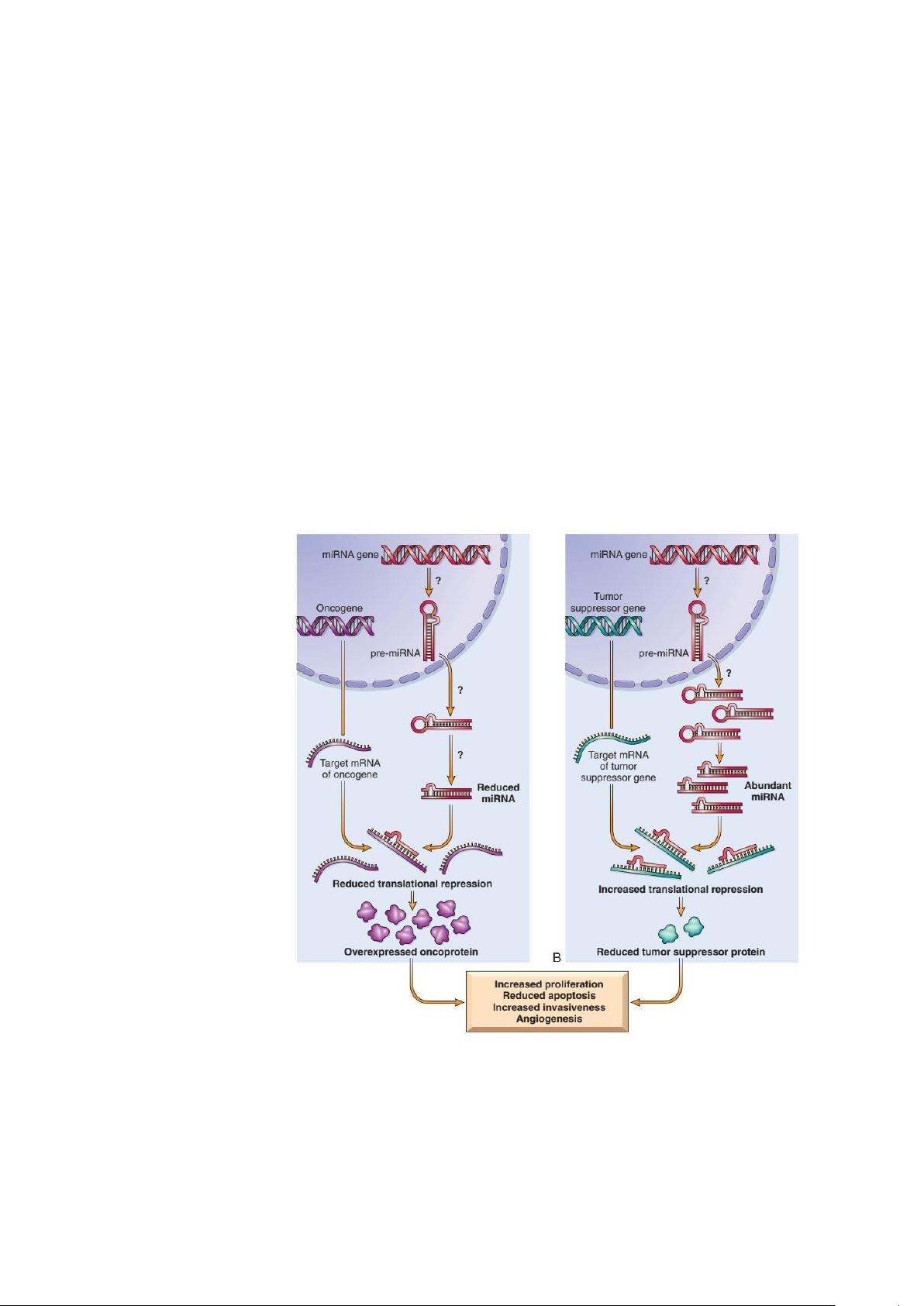
Cơ chế micro RNAs
Micro RNAs (miRNA) là một nhóm các RNA không mã hóa, nội sinh, mạch đơn,
có chiều dài ngắn, chỉ khoảng 21-25 nucleotides. Sự hình thành và hoạt động của miRNA
có thể được tóm tắt qua các bước sau. Đầu tiên các men RNA polymerase II sẽ giúp
phiên mã các gen miRNA để tạo thành các pri-miRNA. Nhờ vào men Drosha, các pri-
miRNA được cắt thành những đoạn pre-miRNA có chiều dài ngắn hơn. Tiếp theo, các
pre-miRNA sẽ được vận chuyển từ trong nhân tế bào ra ngoài bào tương. Tại đây, pre-
miRNA sẽ được cắt thành các miRNA có mạch đôi nhờ vào men Dicer. Sau đó, một
mạch miRNA được lựa chọn và kết hợp với RISC (RNA-inducing silencing complex)
để tạo thành phức hợp RISC/miRNA. Phức hợp RISC/miRNA này sẽ tương tác với đoạn 14 lOMoAR cPSD| 45469857
mRNA đích và gây thoái biến mRNA hay ức chế sự dịch mã. Ước tính cho thấy có
khoảng hơn 30% gen người là đích nhắm của miRNA và miRNA tham gia chức năng
trong hầu hết những quá trình sinh học của cơ thể như điều hòa chu kỳ tế bào, tăng
trưởng tế bào, chết tế bào theo lập trình , biệt hóa tế bào... Vai trò của miRNA trong cơ
chế sinh ung thư có thể chia thành 2 nhóm lớn. Đầu tiên là những miRNA có vai trò sinh
ung thư (còn được gọi là Onco-miR), thường sẽ được tăng biểu hiện để gia tăng tác động
vào các mRNA cùa gen đè nén bướu, gen kiểm soát sự biệt hóa tế bào hoặc chết tế bào
theo lập trình, gây ức chế sự tổng hợp các protein chức năng. Tiếp theo là những miRNA
có vai trò đè nén bướu (còn gọi là Tumor suppressor miR) với đích nhắm là mRNA của
các gen sinh ung. Để ung thư được hình thành thì các miRNA có vai trò đè nén bướu
này cần được giảm biểu hiện để không can thiệp vào quá trình ức chế sự dịch mã những
mRNA của gen sinh ung... Hiện nay, miRNA đang được nghiên cứu nhiều trong lĩnh vực
ung thư như là một dấu ấn sinh học góp phần trong chẩn đoán (chẩn đoán bệnh ở giai
đoạn sớm, phân biệt bướu lành tính và ác tính), tiên đoán (khả năng đáp ứng với các
phương pháp điều trị), theo dõi tái phát và tiên lượng bệnh.
Hình 12: Sự hình thành và vai trò của microRNAs trong sự sinh ung thư
III. KẾT LUẬN
Sự sinh ung thư là tiến trình phức tạp, cần có thời gian để các tế bào tích lũy
những tổn thương di truyền (cơ chế gen) và những phản ứng hóa học, những sự thay đổi
trên chuỗi DNA mà không làm thay đổi cấu trúc của gen (cơ chế ngoài gen). Những rối 15 lOMoAR cPSD| 45469857
loạn của cơ chế gen, ngoài gen chủ yếu là do sự tác động của môi trường bên ngoài (yếu
tố vật lý, hóa học, sinh học) và chỉ một số ít là do di truyền. Ngày nay, với sự tiến bộ về
những kỹ thuật sinh học phân tử đã giúp y học có những hiểu biết sâu hơn, rõ hơn về cơ
chế sinh ung thư. Hy vọng những thành tựu đạt được trong lĩnh vực này sẽ góp phần cải
thiện khả năng chẩn đoán sớm, điều trị, tiên lượng cũng như phòng ngừa bệnh ung thư.
TÀI LIỆU THAM KHẢO
1. Chen Q.W, Zhu X.Y, Li Y.Y, Meng Z.Q.. “Epigenetic regulation and cancer
(review)”. Oncol Rep. 2014 Feb;31(2):523-32.
2. Craig A.A and Sheila A.B. “Cancer: Basic Science and Clinical Aspects, 2010”. Wiley-Blackwell.
3. Kumar V, Abbas A.K, Aster J.C. “Robbins Basic Pathology 9th, 2013”. Elsevier Saunders.
4. Lord C.J, Ashworth A. “The DNA damage response and cancer therapy”. Nature (2012). 481(7381):287-94.
5. https://docplayer.net/10006876-Molecular-basis-of-cancer.html 16
Tài liệu liên quan:
-

Bài giảng sinh học phân tử | Đại học Y Dược Thành phố Hồ Chí Minh
55 28 -

Bài tập sinh di truyền cho môn sinh học phân tử | Đại học Y Dược Thành phố Hồ Chí Minh
60 30 -

Bài 6: Điều Hòa Hoạt Động Gen - Câu Hỏi Sinh Học Phân Tử | Đại học Y Dược Thành phố Hồ Chí Minh
60 30 -

Bài 3 - Các loại ARN: Câu hỏi trắc nghiệm Sinh học phân tử | Đại học Y Dược Thành phố Hồ Chí Minh
61 31 -

Câu hỏi ngắn Sinh học phân tử | Đại học Y Dược Thành phố Hồ Chí Minh
110 55