Đề Cương Ôn Tập | Tin sinh học | Trường Đại học Khoa học Tự nhiên, Đại học Quốc gia Hà Nội
Cơ sở dữ liệu sinh học là thư viện khoa học sinh học, được thu thập từ các thí nghiệm khoa học, tài liệu đã xuất bản, công nghệ thí nghiệm thông lượng cao và phân tích tính toán. Chúng chứa thông tin từ các lĩnh vực nghiên cứu bao gồm gen, proteomics, chuyển hóa, biểu hiện gen microarray và phát sinh loài. Tài liệu được sưu tầm và soạn thảo dưới dạng file PDF để gửi tới các bạn cùng tham khảo, ôn tập đầy đủ kiến thức, chuẩn bị cho các buổi học thật tốt. Mời bạn đọc đón xem!
Môn: Tin sinh học (HUS) 10 tài liệu
Trường: Trường Đại học Khoa học tự nhiên, Đại học Quốc gia Hà Nội 1.1 K tài liệu
Tác giả:


















Preview text:
TRƯỜNG ĐẠI HỌC KHOA HỌC TỰ NHIÊN – ĐHQGHN KHOA SINH HỌC o0o VŨ HẢI DƯƠNG K64 SINH HỌC
ĐỀ CƯƠNG ÔN TẬP HỌC PHẦN
TIN SINH HỌC – BIOINFORMATICS
Mã học phần: BIO2220 – Học kỳ II năm 2021 – 2022
Hà Nội – Tháng 06/2022 2 MỤC LỤC
Phần 1. CƠ SỞ DỮ LIỆU SINH HỌC ........................................................................................ 4
1. Giới thiệu chung về cơ sở dữ liệu sinh học .......................................................................... 4
2. Các khái niệm trong tin sinh học ......................................................................................... 4
3. Cơ sở dữ liệu NCBI - National Center for Biotechnology Information .............................. 5
4. Một số cơ sở dữ liệu và công cụ genome và protein .......................................................... 6
Phần 2. SO SÁNH TRÌNH TỰ SINH HỌC ............................................................................... 8
1. So sánh trình tự sinh học ...................................................................................................... 8
2. Phương pháp so sánh trình tự............................................................................................... 8
a. Phương thức so sánh ......................................................................................................... 8
b. Ma trận so sánh ................................................................................................................. 9
3. So sánh hai trình tự (pairwise) ........................................................................................... 11
a. So sánh ma trận điểm ...................................................................................................... 11
b. Lập trình động .................................................................................................................. 11
c. BLAST ............................................................................................................................ 11
4. So sánh nhiều trình tự ........................................................................................................ 11
Phần 3. PHÂN TÍCH CÂY CHỦNG LOẠI PHÁT SINH ...................................................... 13
1. Phân tích chủng loại phát sinh ........................................................................................... 13
2. Phương pháp xây dựng cây chủng loại phát sinh .............................................................. 13
3. Cây phát sinh chủng loại phát sinh trên toàn bộ genome .................................................. 15
Phần 4. DỰ ĐOÁN GENE ......................................................................................................... 16
1. Dự đoán và chú giải gen .................................................................................................... 17
2. Phương pháp dự đoán và chú giải gen ............................................................................... 17
Phần 5. MOTIF VÀ DOMAIN CỦA DNA VÀ PROTEIN .................................................... 18
1. Motif và Domain ................................................................................................................ 18
2. Các phương pháp tìm kiếm motif và domain (module) ..................................................... 18
a. Phương pháp liệt kê ........................................................................................................ 18
b. Phương pháp tối ưu hóa xác định ................................................................................... 19
c. Phương pháp tối ưu hóa xác suất .................................................................................... 19
3. Protein – dự đoán cấu trúc và chức năng ........................................................................... 19
Phần 6. THIẾT KẾ MỒI CHO PHẢN ỨNG PCR .................................................................. 21
1. Tiêu chuẩn trong việc thiết kế mồi .................................................................................... 21
a. Mục đích của mồi ........................................................................................................... 21
b. Độ dài của mồi và sản phẩm khuếch đại ........................................................................ 21
c. Nhiệt độ nóng chảy của mồi ........................................................................................... 22
d. Hàm lượng GC trong mồi ............................................................................................... 22 2 3
e. Nhiệt độ gắn mồi ............................................................................................................ 22
Phần 7. NETWORK SINH HỌC .............................................................................................. 23
1. Network là gì? .................................................................................................................... 23
2. Các yếu tố cần quan tâm trong một Network .................................................................... 23
a. Distance .......................................................................................................................... 23
b. Average Path length (L) .................................................................................................. 23
c. Diameter ......................................................................................................................... 24
d. Degree (k) ....................................................................................................................... 24
e. Hệ số nhóm (Clustering coefficient) ............................................................................... 25
f. Hubs ................................................................................................................................ 25
3. Tương tác protein – protein ............................................................................................... 25
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2011 – 2012 ........................................... 28
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2012 – 2013 ........................................... 28
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2014 – 2015 ........................................... 28
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2015 – 2016 ........................................... 29
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2016 – 2017 ........................................... 29
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2019 – 2020 ........................................... 29
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2020 – 2021 (K64CLC) ........................ 30
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2020 – 2021 (K63CNSH) ...................... 30
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ I NĂM 2020 – 2021 (K63CLC và K62CNSH) .. 31
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ I NĂM 2021 – 2022 ............................................ 32
ĐỀ THI KẾT THÚC HỌC PHẦN – KỲ II NĂM 2021 – 2022 ........................................... 33 3 4
Phần 1. CƠ SỞ DỮ LIỆU SINH HỌC
1. Giới thiệu chung về cơ sở dữ liệu sinh học
Cơ sở dữ liệu sinh học là thư viện khoa học sinh học, được thu thập từ các thí nghiệm
khoa học, tài liệu đã xuất bản, công nghệ thí nghiệm thông lượng cao và phân tích tính
toán. Chúng chứa thông tin từ các lĩnh vực nghiên cứu bao gồm gen, proteomics, chuyển
hóa, biểu hiện gen microarray và phát sinh loài.
Cơ sở dữ liệu sinh học được sử dụng dưới dạng những phần mềm, mục đích hướng tới
của các phần mềm này là dễ sử dụng, giao diện đẹp và chất lượng kết quả tốt. Trong thực tế có
hai dạng các chương trình công cụ ứng dụng tin sinh học: phần mềm sử dụng trực tuyến trên
internet và phần mềm yêu cầu tải về máy. Cả hai dạng này có thể là miễn phí nhưng cũng có thể yêu cầu phải trả phí.
Trong một phần mềm ứng dụng có thể bao gồm rất nhiều các phần mềm nhỏ hơn, ví dụ
như cơ sở dữ liệu và máy chủ ExPASy (https://www.expasy.org/)... Hay có những phần mềm
được sử dụng là dữ liệu gốc sử dụng cho các chương trình khác, ví dụ như cơ sở dữ liệu về
protein Uniprot (http://www.uniprot.org/), cơ sở dữ liệu NCBI (https://www.ncbi.nlm.nih.gov/) …
2. Các khái niệm trong tin sinh học
Trình tự là một tập hợp các ký tự tuyến tính (các phần tử của trình tự) đại diện cho các
nucleotide (DNA, RNA) hay amino acid (protein). Việc biểu diễn các trình tự này trong quá trình
xử lý và phân tích tin sinh học cần sự đơn giản, dễ hiểu và ngắn gọn, hạn chế tối ưu dung lượng
bộ nhớ, do đó với DNA/RNA bao gồm nucleotide là A, T/U, G, C; protein bao gồm 20 loại ký
hiệu khác nhau là một chữ cái duy nhất cho từng amino acid.
Tuy vậy trong thực tế, việc nghiên cứu có thể có một vài vị trí trong trình tự mà ta không
chắc chắn nó là loại nào. Do đó ta cũng có những ký hiệu riêng biệt cho những sự không chắc
chắn đó. Những quy ước sử dụng trong Tin sinh học, được Hiệp hội Hóa sinh học Quốc tế đề
xuất và thông qua để sử dụng gọi là mã IUB (DNA) và mã SLC (protein):
• Với nucleic acid sử dụng: A, C, G, T, U: • Với protein sử dụng:
Định dạng các file trình tự trong quá trình có thể chia làm các loại: 4 5
- File text hay nhị phân: File text dùng mã IUB và có thể đọc được bằng word processer
(simple text, Microsoft Word) hay text editor (emacs). File nhị phân thường chỉ được đọc
bằng chương trình tạo ra nó (MacVector).
- Tối thiểu (FASTA) hay chú thích (GCG): định dạng tối thiểu dành cho những trình tự
nucleotide hay peptide biểu diễn bằng một chữ, bắt đầu bằng một dòng miêu tả (>name)
sau đó là dòng trình tự. Định dạng chủ thích chứa chính xác một trình tự bắt đầu với một
dòng chú thích và bắt đầu trình tự được đánh dấu bởi một dòng kết thúc bằng “..” dòng
trước trình tự chứa các mã nhận diện, độ dài trình tự.
3. Cơ sở dữ liệu NCBI - National Center for Biotechnology Information
Một trong số các cơ sở dữ liệu miễn phí có uy tín là NCBI. Cơ sở dữ liệu NCBI do Trung
tâm Thông tin Công nghệ Sinh học Quốc gia Hoa Kỳ và Đại học Y Quốc gia Hoa Kỳ hợp tác
cung cấp từ 1988; cho phép người dùng sử dụng miễn phí để tra cứu, tham khảo các tài liệu là
sách, bài báo khoa học chủ yếu thuộc lĩnh vực công nghệ sinh học, y sinh. Người dùng có thể tìm
kiếm tất cả các bộ sưu tập dữ liệu hoặc tìm kiếm trong từng bộ sưu tập dữ liệu thành phần như
PubMed, PubMed Central, PubChem, Genome, …
Entrez là công cụ tìm kiếm nhanh, thống kê kết quả một cách đầy đủ và khoa học được
NCBI sử dụng. Sử dụng entrez có thể tìm kiếm:
Pubmed: Tài liệu, bài báo, tạp chí, sách,… liên quan đến lĩnh vực sinh y
Genbank: Cơ sở dữ liệu trình tự nucleotide
Protein sequence database: Cơ sở dữ liệu trình tự protein
SNP: đa hình đơn nucleotide
PopSet: bộ dữ liệu nghiên cứu về dân số
Taxonomy: phân loại sinh vật dựa trên Genbank ….
Trang chủ của cơ sở dữ liệu NCBI. Để sử dụng Entrez gõ từ khóa vào thanh tìm kiếm, chọn
loại dữ liệu ở ô bên trái và nhấn Search để tìm kiếm
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC
Sơ đồ khối về quá trình hoạt động của công cụ t 6 ì m kiếm Entrez:
4. Một số cơ sở dữ liệu và công cụ genome và protein
ExPASy là một cổng thông tin tin sinh học của viện tin sinh Thụy Sĩ (SIB) từ 2011. Đặc
biệt, ExPASy được thiết kế, phát triển và duy trì bởi SIB Web Team trong sự cộng tác với một số
nhóm SIB và khác và người dùng ExPASy. ExPASy là một cổng thông tin rộng và tích hợp truy
cập nhiều tài nguyên khoa học, cơ sở dữ liệu và công cụ phần mềm ở các lĩnh vực của khoa học
(Proteomics, genomics, transcriptomics, chủng loại phát sinh, sinh học hệ thống, tiến hóa, di
truyền dân số). ExPASy là viết tắt của Expert Protein Analysis System UniProt (Universal
Protein resourse) là một tài nguyên toàn diện cho dữ liệu protein và dữ liệu chú thích.
UniProt gồm UniProt Knowledgebase (UniProtKB), UniProt Reference Clusters
(UniRef), và UniProt Archive (UniParc). UniProt là sự hợp tác giữa Viện Thông tin Sinh học
Châu Âu (EMBL-EBI), Viện Thông tin Sinh học Thụy Sĩ (SIB) và nguồn thông tin về Protein
(PIR). Năm 2002, ba viện nghiên cứu đã quyết định tập hợp các nguồn lực và chuyên môn của
họ và thành lập liên minh UniProt. NCBI ( National Center for Biotechnology Information) của
Mỹ là một bộ phận của NIH. cơ sở dữ liệu của NCBI rất rộng bao gồm nhiều mảng: văn hóa
phẩm khoa học (sách, tạp chí và công bố khoa học), sức khỏe, DL về genome, Genes, proteins,
hóa chất… ngoài ra là các công cụ phân tích
EBI (European Bioinformatics Institute) cung cấp dữ liệu sinh học công bố trên toàn thế
giới cho cộng đồng khoa học miễn phí thông qua nhiều dịch vụ và công cụ, thực hiện nghiên cứu
cơ bản và đào tạo chuyên nghiệp về tin sinh học. EBI là thành viên của Phòng thí nghiệm sinh
học phân tử châu Âu (EMBL), một tổ chức nghiên cứu quốc tế, sáng tạo và liên ngành được tài
trợ bởi 22 quốc gia thành viên và 2 quốc gia thành viên liên kết.
DDBJ (DNA Data Bank of Japan) thu thập dữ liệu trình tự nucleotide như một thành
viên của INSDC (International Nucleotide Sequence Database Collaboration) và cung cấp dữ
liệu trình tự nucleotide sẵn có và hệ thống siêu máy tính, để hỗ trợ các hoạt động nghiên cứu
trong khoa học sự sống. Vì Trung tâm DDBJ trao đổi dữ liệu được công bố với ENA/ EBI và
NCBI hàng ngày, ba trung tâm dữ liệu chia sẻ hầu như cùng một dữ liệu tại bất kỳ thời điểm nào.
Ngoài ra còn có các chương trình GenomeNet, EMBNet, PDB,…
BLAST trong NCBI, BLAST tìm các vùng giống nhau giữa các trình tự sinh học.
Chương trình so sánh trình tự nucleotide hoặc protein với cơ sở dữ liệu trình tự và tính toán ý nghĩa thống kê.
CLustal là một loạt các chương trình máy tính được sử dụng rộng rãi được sử dụng
trong tin sinh học để căn chỉnh nhiều trình tự. Đã có nhiều phiên bản của Clustal về sự phát triển
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 7
của thuật toán như Clustal, ClustalV, ClustalW, ClustalX, Clustal Omega, Clustal2. Phiên bản
gần đây hơn của phần mềm có sẵn cho Windows, Mac OS và Unix / Linux. Nó cũng thường
được sử dụng thông qua một giao diện web tại trang chủ Clustal hoặc được sử dụng qua trang
chủ của Viện Tin sinh học Châu Âu.
Treeview là một chương trình để hiển thị và in tệp phân tích phát sinh loài. Chương trình
đọc hầu hết các tệp cây NEXUS (chẳng hạn như các tệp được sản xuất bởi PAUP và
COMPONENT) và các tệp cây kiểu PHYLIP (bao gồm cả các tệp được sản xuất bởi fastDNAml và CLUSTALW).
SWISS-MODEL là một máy chủ mô hình hóa cấu trúc protein hoàn toàn tự động, có thể
truy cập qua máy chủ web ExPASy, hoặc từ chương trình DeepView (Swiss Pdb-Viewer). Mục
đích của máy chủ này là làm cho Protein Modeling có thể tiếp cận được với tất cả các nhà sinh
học và các nhà sinh học phân tử trên toàn thế giới. Người dùng chỉ việc đưa trình tự lên với định
dạng Fasta, Clustal, Promod, plain string, …
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 8
Phần 2. SO SÁNH TRÌNH TỰ SINH HỌC
1. So sánh trình tự sinh học
So sánh các trình tự sinh
học có mục đích là tìm ra các
thông tin, mối liên hệ về cấu trúc
từ đó suy luận về chức năng và quá
trình tiến hóa. Các trình tự tương
đồng có thể thực hiện các chức
năng tương đồng nhau, giúp điều
hòa biểu hiện gene, các chức năng
hóa sinh học, tạo thành các tương đồng về cấu trúc, chỉ ra nguồn gốc chung, tổ tiên chung của các loài.
So sánh trình tự của DNA, RNA hay protein để xác định các vùng giống nhau có thể là
hệ quả của mối quan hệ về chức năng, cấu trúc và tiến hóa giữa các trình tự. Nếu hai trình tự
trong một phép so sánh có nguồn gốc chung thì các điểm không khớp (mismatch) được giải thích
là đột biến điểm, các khoảng trống (gap) xuất hiện trong một hay cả hai trình tự tại thời điểm
chúng phân ly khỏi nhau trong quá trình tiến hóa. Trong so sánh trình tự protein, mức độ giống
nhau của các amino acid ở vị trí đặc biệt trong trình tự có thể gợi ý về sự bảo tồn một vùng nhất
định hay motif giữa các loài. Nếu một vùng rất bảo thủ chứng tỏ rằng vùng này có một cấu trúc
và chức năng vô cùng quan trọng.
2. Phương pháp so sánh trình tự
Các trình tự rất ngắn và giống nhau có thể thực hiện bằng các phương pháp thủ công. Với
các phương pháp dựa vào máy tính để so sánh trình thường rơi vào hai nhóm: so sánh toàn thể và so sánh cục bộ.
a. Phương thức so sánh
So sánh trình tự toàn thể (global alignment)
So sánh trình tự toàn thể
là một dạng của tối ưu hóa toàn
diện, các phép so sánh phải triển
khai trên toàn bộ độ dài của các
trình tự được so sánh. So sánh
này bắt đầu ở hai trình tự và thêm
các gap vào mỗi trình tự cho đến
khi chạm đến tận cùng mỗi trình tự. Thuật toán sử dụng là Needleman – Wunsh.
So sánh trình tự cục bộ (local alignment)
So sánh trình tự cục bộ xác định các vùng giống nhau nhất giữa hai trình tự và xây dựng
phép so sánh kể từ đó. So sánh cục bộ thường được ưu tiên hơn, nhưng có thể khó khăn trong
tính toán vì phải xác định các vùng tương đồng. Hữu ích hơn đối với các trình tự không giống
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 9
nhau được nghi ngờ là chứa vùng giống nhau hay motif giống nhau trong cả trình tự lớn. Thuật
toán được sử dụng là Smith – Waterman.
Các thuật toán so sánh có các thông số để kiểm tra độ chính xác của kết quả. Hệ thống
tính điểm là một thông số quan trọng. Mỗi cặp biểu tượng được gán cho một giá trị số, dựa vào
một bảng so sánh các biểu tượng. Hệ thống tính điểm DNA rất đơn giản với match = 1 và
mismatch = 0 tạo thành các ma trận nhị phân. Hệ thống tính điểm cho protein các sự phức tạp
hơn, các amino acid có đặc tính hóa sinh và lý sinh khác nhau ảnh hưởng tới khả năng có thể
thay thế của chúng trong quá trình tiến hóa. Hệ thống tính điểm protein phản ánh thứ tự các đột
biến để chuyển amino acid này thành amino acid khác, tính tương tự về mặt hóa học, tần số đột
biến quan sát được, xác suất xuất hiện mỗi loại amino acid. b. Ma trận so sánh
Ma trận có thể được sử dụng để tính điểm gồm ma trận nhị phân, ma trận đặc hiệu vị trí
(PSSM) và ma trận thống kê. Hai ma trận thống kê thường được sử dụng là PAM và BLOSUM.
Ma trận PAM (Percent Accepted Mutations) Ma trận này được tính bằng cách quan
sát sự khác biệt trong các protein liên quan chặt chẽ. Bởi vì việc sử dụng các homologs liên quan
rất chặt chẽ, các đột biến quan sát được dự kiến sẽ không thay đổi đáng kể các chức năng phổ
biến của các protein. Do đó, các thay thế quan sát được (theo đột biến điểm) được coi là được
chấp nhận bởi chọn lọc tự nhiên. Một đơn vị PAM được định nghĩa là 1% các vị trí axit amin đã
được thay đổi. Để tạo ra một ma trận thay thế PAM1, một nhóm các chuỗi liên quan rất chặt chẽ
với tần số đột biến tương ứng với một đơn vị PAM được chọn. Dựa trên dữ liệu đột biến được
thu thập từ nhóm trình tự này, một ma trận thay thế có thể được bắt nguồn. Ma trận PAM1 này
ước tính tỷ lệ thay thế sẽ được dự kiến nếu 1% axit amin đã thay đổi. Ma trận PAM1 được sử
dụng làm cơ sở để tính toán các ma trận khác bằng cách giả định rằng các đột biến lặp đi lặp lại
sẽ theo mô hình tương tự như trong ma trận PAM1 và nhiều sự thay thế có thể xảy ra tại cùng
một vị trí. Sử dụng logic này, Dayhoff có nguồn gốc ma trận cao như PAM250. Thông
thường PAM 30 và PAM70 được sử dụng. Vai trò của ma trận PAM là xuất phát từ so sánh toàn
thể các họ protein, các thành viên của họ chung có tối thiểu 85% điểm tương đồng, giúp xây
dựng cây chủng loại phát sinh và các trình tự tổ tiên của mỗi họ protei và ước tính số thay thế đối với mỗi cặp amino acid. Hạn chế của ma trận PAM là nó chỉ dựa vào
một bộ dữ liệu gốc, kiểm
tra các protein với ít khác
biệt (tương đồng >85%)
và chủ yếu nó dựa vào
các protein dạng cầu nhỏ
vì thế ma trận này tính khách quan chưa cao.
Ma trận BLOSUM (Blocks Substituation Matrix) ma trận này sử dụng nhiều liên kết
của các protein phân kỳ tiến hóa. Các xác suất được sử dụng trong tính toán ma trận được tính
toán bằng cách xem xét các "khối" của các chuỗi được bảo tồn được tìm thấy trong nhiều liên kết
protein. Các chuỗi được bảo tồn này được cho là có tầm quan trọng chức năng trong các protein
liên quan và do đó sẽ có tỷ lệ thay thế thấp hơn so với các khu vực ít được bảo tồn hơn. Để giảm
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 10
sự thiên vị từ các chuỗi liên quan chặt chẽ về tỷ lệ thay thế, các phân đoạn trong một khối có
nhận dạng trình tự trên một ngưỡng nhất định đã được nhóm lại, làm giảm trọng lượng của mỗi
cụm như vậy. Đối với ma trận BLOSUM62, ngưỡng này được đặt ở mức 62%. Tần số cặp sau đó
được tính giữa các cụm, do đó các cặp chỉ được tính giữa các phân đoạn dưới 62% giống hệt
nhau. Người ta sẽ sử dụng ma trận BLOSUM được đánh số cao hơn để căn chỉnh hai chuỗi liên
quan chặt chẽ và số thấp hơn cho các chuỗi phân kỳ hơn.
Nó chỉ ra rằng ma trận BLOSUM62 thực hiện một công việc tuyệt vời phát hiện sự tương đồng
trong các chuỗi xa, và đây là ma trận được sử dụng theo mặc định trong hầu hết các ứng dụng
căn chỉnh gần đây như BLAST. Xuất phát từ các phép so sánh các domain của các protein có
quan hệ xa. Những sự xuất hiện các cặp amino acid trên mỗi cột của mỗi khối so sánh sẽ được
đếm.. Những số có nguồn gốc từ tất cả các khối được dùng để tính ma trận BLOSUM. Các trình
tự trong các khối Block được nhóm lại theo mức độ tương đồng của chúng và được tính toán như
là một trình tự duy nhất. Số n (BLOSUMn) thể hiện phần trăm tương đồng trình tự được dùng để
xây dựng ma trận, số càng lớn thì khoảng cách tiến hóa càng nhỏ. Thông thường thì
BLOSUM sẽ tốt hơn PAM với các
tìm kiếm tương đồng cục bộ. Khi
so sánh các protein có quan hệ gần
gũi nên dùng ma trận PAM hoặc
ma trận BLOSUM có chỉ số cao,
đối với protein có khoảng cách xa
nhau nên dùng PAM cao hơn và
BLOSUM thấp hơn. Đối với việc
rà soát cơ sở dữ liệu, thường hay sử dụng BLOSUM62. Với việc so sánh tương
đồng protein, người ta quan tâm
tới sự đóng góp (%) của các vị trí
tương đồng, độ dài của chuỗi so
sánh, sự phân bố các vị trí tương
đồng dọc theo trình tự, các gốc ở
vị trí bảo thủ và tính tương đồng
về cấu trúc, di truyền của amino
acid ở các vị trí không tương đồng.
Mô hình Markov cho rằng xác suất thay thế amino acid này bằng amino acid khác là như
nhau, bất kể amino acid nào được chuyển thành từ amino acid nào. Những đột biến thay thế
amino acid dựa vào một lần chuyển duy nhất không tuân theo mô hình Markov thì xác suất thay
thế một amino acid này thành amino acid khác phụ thuộc đáng kể vào là amino acid nào đã
chiếm giữ vị trí đó trong quá khứ. Phần nào của codon chứa đựng thông tin về amino acid trước
đó đã xuất hiện ở vị trí nào đó của trình tự protein. Thông tin về codons của amino acid trước đó
được lưu giữa trong thời gian nào.
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 11
3. So sánh hai trình tự (pairwise)
Các phương pháp so sánh hai trình tự được dùng để tìm ra những phần trùng khớp nhất
(cục bộ) hoặc so sánh toàn thể hai trình tự được truy vấn. Chỉ có thể được dùng giữa hai trình tự
cùng lúc nhưng lại hiệu quả trong quá trình tính toán và thường được dùng cho các phương pháp
không yêu cầu độ chính xác cao. Có 3 phương pháp để so sánh hai trình tự:
a. So sánh ma trận điểm
Ma trận điểm (dot-matrix) tạo ra các phép so sánh giữa các vùng trình tự riêng lẻ, là định
tính và đơn giản về mặt nhận thức mặc dù tốn thời gian phân tích trên quy mô lớn. Nó dễ dàng
có thể quan sát một số đặc điểm như việc lặp lại, mất, thêm, đảo chiều từ một biểu đồ ma trận
điểm. Kích thước và mật độ của điểm phụ thuộc vào mức độ giống nhau của hai trình tự. Các
điểm biểu diễn của hai trình tự liên hệ rất gần nhau sẽ xuất hiện như một đường thẳng dọc theo
đường chéo chính của ma trận.
*Nhược điểm: gây nhiễu rất nhiều và không rõ ràng, thiếu tính trực quan, khó có thể thu thập số
liệu thống kê tóm tắt của hai trình tự, lãng phí không gian so sánh
*Ưu điểm: Còn có thể đánh giá sự lặp lại của một chuỗi, dành cho các protein đa tiểu phần có các domain tương đồng
b. Lập trình động
Đây là một phương pháp lập trình phổ biến, có thể áp dụng khi không gian tìm kiếm lớn
có thể xây dựng thành một loạt các bước: (1) chia thành nhiều vấn đề nhỏ (2) giải quyết từng
bước bằng các giải pháp nhỏ (3) kết hợp các kết quả để tìm giải pháp cuối.
So sánh protein sử dụng ma trận thay thế để gán số điểm cho những amino acid trùng
khớp hay không trùng, một gap cho một amino acid có trên trình tự này những không có trên trình tự kia.
So sánh DNA hay RNA có thể sử dụng một ma trận tính điểm, thực tế thường gán một
điểm cộng trùng khớp với một điểm trừ không trùng khớp, và một điểm trừ gap.
Phương pháp này hữu ích trong việc sắp xếp các trình tự nucleotide thành protein. c. BLAST
BLAST (Basic Local Aligment Search Tool) tìm kiếm các vùng tương đồng cục bộ giữa
các trình tự. Chương trình so sánh các trình tự nucleotide hay protein với cơ sở dữ liệu và tính
toán ra mức độ trùng khớp có ý nghĩa về mặt thống kê. BLAST được dùng để kết luận mối quan
hệ giữa chức năng và tiến hóa giữa các trình tự cũng như xác định các thành viên trong cùng một họ.
Phương pháp này sử dụng thuật toán Smith – Waterman, là công cụ tìm kiếm tương đồng
được sử dụng rất nhiều vì nó tìm ra sự so sánh cục bộ tốt nhất, cung cấp ý nghĩa về mặt thống kê.
BLAST hoạt động bawfg cách tìm kiếm những trùng khớp về word giữa trình tự truy xuất và cơ sở dữ liệu.
4. So sánh nhiều trình tự
So sánh nhiều trình tự là phép so sánh nhiều hơn hai trình tự thu được bằng việc chèn gap
vào trình tự vì thế làm cho các trình tự có độ dài bằng nhau và có thể so dánh trong một ma trận.
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 12
Nguyên lý cơ bản của so sánh nhiều trình tự là việc so sánh liên tục hai trình tự theo từng
cặp. Mục đích của việc so sánh này có thể xác định được các họ protein, vùng tương đồng chung
trong một bộ so sánh, xác định trịnh tự liên ứng của một số trình tự, chỉ ra những cấu trúc bậc cao hơn của proteim.
Có 3 phương pháp để so sánh nhiều trình tự:
Lập trình động (dymanic programming approach) tính toán một phép so sánh tối ưu
cho một hàm tính điểm cho trước, nhược điểm là thời gian chạy lâu nên ít người sử dụng Phương pháp tiếp
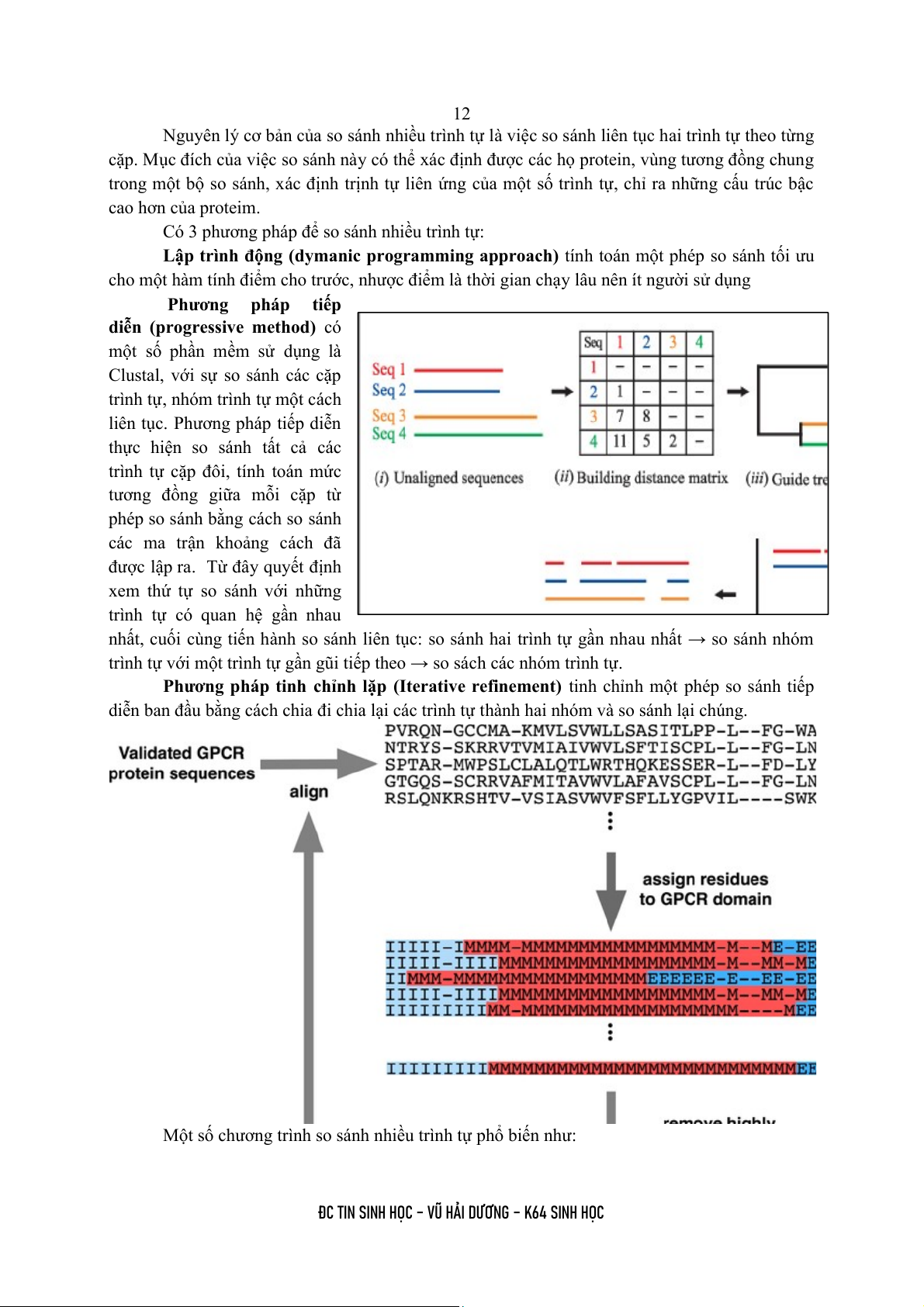
diễn (progressive method) có
một số phần mềm sử dụng là
Clustal, với sự so sánh các cặp
trình tự, nhóm trình tự một cách
liên tục. Phương pháp tiếp diễn
thực hiện so sánh tất cả các
trình tự cặp đôi, tính toán mức
tương đồng giữa mỗi cặp từ
phép so sánh bằng cách so sánh
các ma trận khoảng cách đã
được lập ra. Từ đây quyết định
xem thứ tự so sánh với những
trình tự có quan hệ gần nhau
nhất, cuối cùng tiến hành so sánh liên tục: so sánh hai trình tự gần nhau nhất → so sánh nhóm
trình tự với một trình tự gần gũi tiếp theo → so sách các nhóm trình tự.
Phương pháp tinh chỉnh lặp (Iterative refinement) tinh chỉnh một phép so sánh tiếp
diễn ban đầu bằng cách chia đi chia lại các trình tự thành hai nhóm và so sánh lại chúng.
Một số chương trình so sánh nhiều trình tự phổ biến như:
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 13
COBALT: tính toán sự sắp xếp trình tự nhiều protein bằng cách sử dụng miền bảo tồn và thông
tin tương tự trình tự cục bộ.
Clustal Omega: Căn chỉnh trình tự và cấu trúc với Tcoffee. Căn chỉnh chính xác hơn với nhược
điểm là thời gian chạy lâu hơn một chút
Phần 3. PHÂN TÍCH CÂY CHỦNG LOẠI PHÁT SINH
1. Phân tích chủng loại phát sinh
Nghiên cứu chủng loại phát sinh là nghiên cứu lịch sử tiến hóa và mối quan hệ giữa các
cá thể hoặc nhóm sinh vật (loài, quần thể). Một cây chủng loại phát sinh là một mô hình giả
thuyết về lịch sử các mối quan hệ tiến hóa của một nhóm sinh vật. Cây chủng loại phát sinh bao
gồm các nhánh và các node. Nhánh kết nối các node, một node biểu thị vị trí chia ra làm các
nhánh. Gốc (root) là điểm cổ xưa nhất của cây. Cây gene là một mô hình về sự tiến hóa của một
gene thông qua lặp lại, mất hay thay thế nucleotide, phản ánh mối quan hệ tiến hóa của nhóm
gene. Cây loài phản ánh mô hình phân ly của các nòi thông quan quá trình phát sinh loài, dựa
vào một gene nào đó giữa các taxon.
Đột biến và phát sinh loài mới không được dự đoán là xảy ra đồng thời, nên cây gene
không thể hiện cho loài. Cây phát sinh chủng loại phân tử có thể có độ dài nhánh tương ứng với
khoảng cách tiến hóa giữa hai node mà nó kết nối. Có hai kiểu biểu diễn cây, cây huyết thống
(phylogram) thể hiện cả mối quan hệ tiến hóa và thông tin về thời gian phân ly khỏi nhau của
nhánh; biểu đồ phân nhánh huyết thống (cladogram) chỉ nói lên mối quan hệ chứ không phản
ánh được khoảng cách tiến hóa.
2. Phương pháp xây dựng cây chủng loại phát sinh
Để xây dựng cây chủng loại phát sinh cần thực hiện: (1) xem xét một nhóm các trình tự
để phân tích (2) so sánh các trình tự này (3) áp dụng các phương pháp để xây dựng cây (4) đánh
giá về mặt thống kê đối với cây được dựng. Để xây dựng cây chủng loại phát sinh cần áp dụng
các phương pháp so sánh nhiều trình tự và kiểm tra bằng bootstraping.
So sánh trình tự cần chính xác và đáng tin cậy, các vị trí gap đều sẽ được bỏ qua. Bước
đầu tiên là cần tạo ra một ma trận khoảng cách giữa hai trình tự vào tất cả các phép so sánh hai
trình tự, sử dụng phương pháp dựa trên thống kê để xây dựng một cây ban đầu. Sau đó sắp xếp
lại dần dần các trình tự theo mức độ liên hệ của chúng so với cây phỏng đoán. Từ phép so sánh
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 14
nhiều trình tự xây dựng một cây mới dựa vào khoảng cách của mỗi cặp. Tiến hành lặp lại quá
trình cho đến khi cây sau giống cây trước.
Có hai nguyên lý để xây dựng cây chủng loại phát sinh:
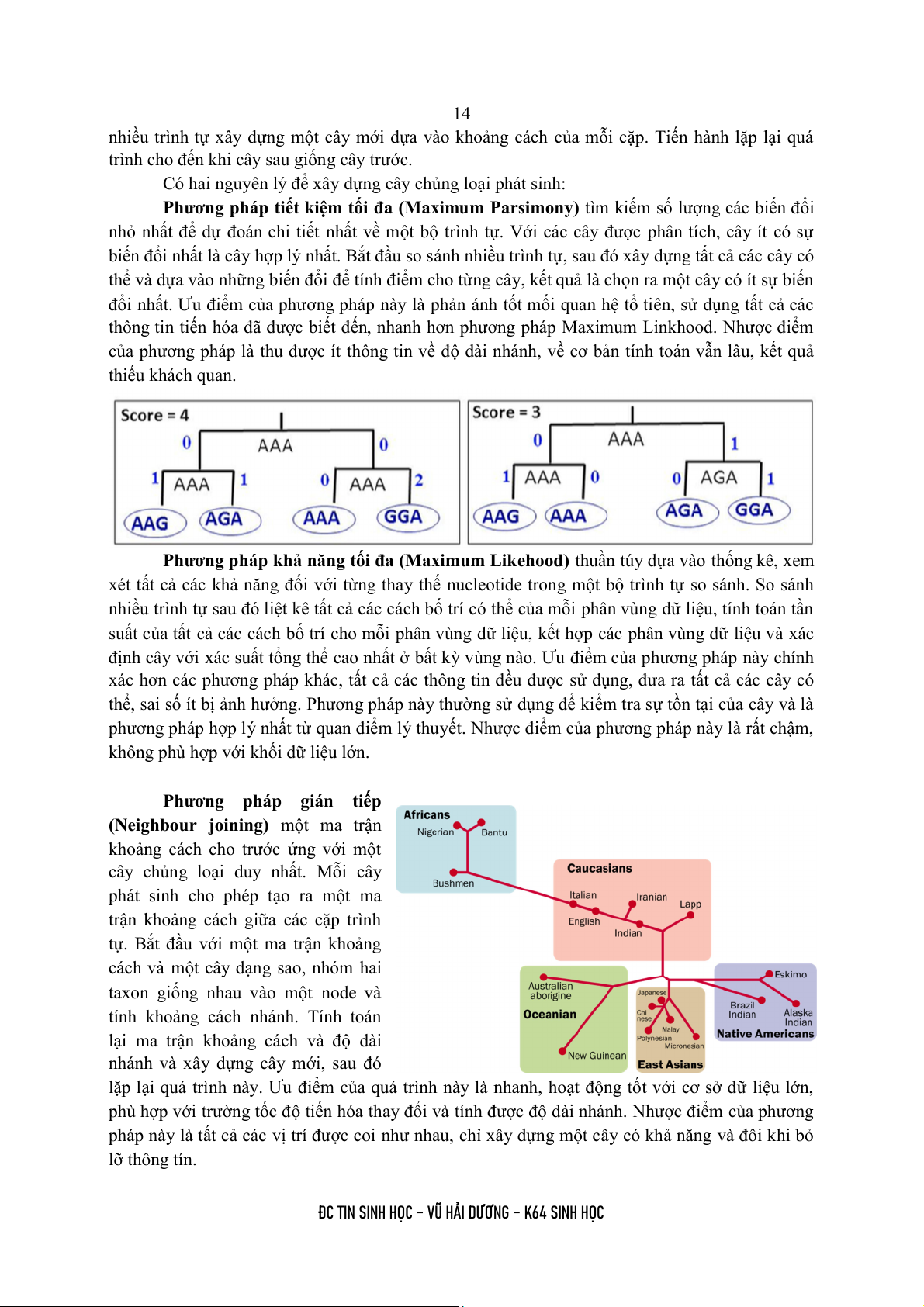
Phương pháp tiết kiệm tối đa (Maximum Parsimony) tìm kiếm số lượng các biến đổi
nhỏ nhất để dự đoán chi tiết nhất về một bộ trình tự. Với các cây được phân tích, cây ít có sự
biến đổi nhất là cây hợp lý nhất. Bắt đầu so sánh nhiều trình tự, sau đó xây dựng tất cả các cây có
thể và dựa vào những biến đổi để tính điểm cho từng cây, kết quả là chọn ra một cây có ít sự biến
đổi nhất. Ưu điểm của phương pháp này là phản ánh tốt mối quan hệ tổ tiên, sử dụng tất cả các
thông tin tiến hóa đã được biết đến, nhanh hơn phương pháp Maximum Linkhood. Nhược điểm
của phương pháp là thu được ít thông tin về độ dài nhánh, về cơ bản tính toán vẫn lâu, kết quả thiếu khách quan.
Phương pháp khả năng tối đa (Maximum Likehood) thuần túy dựa vào thống kê, xem
xét tất cả các khả năng đối với từng thay thế nucleotide trong một bộ trình tự so sánh. So sánh
nhiều trình tự sau đó liệt kê tất cả các cách bố trí có thể của mỗi phân vùng dữ liệu, tính toán tần
suất của tất cả các cách bố trí cho mỗi phân vùng dữ liệu, kết hợp các phân vùng dữ liệu và xác
định cây với xác suất tổng thể cao nhất ở bất kỳ vùng nào. Ưu điểm của phương pháp này chính
xác hơn các phương pháp khác, tất cả các thông tin đều được sử dụng, đưa ra tất cả các cây có
thể, sai số ít bị ảnh hưởng. Phương pháp này thường sử dụng để kiểm tra sự tồn tại của cây và là
phương pháp hợp lý nhất từ quan điểm lý thuyết. Nhược điểm của phương pháp này là rất chậm,
không phù hợp với khối dữ liệu lớn.
Phương pháp gián tiếp
(Neighbour joining) một ma trận
khoảng cách cho trước ứng với một
cây chủng loại duy nhất. Mỗi cây
phát sinh cho phép tạo ra một ma
trận khoảng cách giữa các cặp trình
tự. Bắt đầu với một ma trận khoảng
cách và một cây dạng sao, nhóm hai
taxon giống nhau vào một node và
tính khoảng cách nhánh. Tính toán
lại ma trận khoảng cách và độ dài
nhánh và xây dựng cây mới, sau đó
lặp lại quá trình này. Ưu điểm của quá trình này là nhanh, hoạt động tốt với cơ sở dữ liệu lớn,
phù hợp với trường tốc độ tiến hóa thay đổi và tính được độ dài nhánh. Nhược điểm của phương
pháp này là tất cả các vị trí được coi như nhau, chỉ xây dựng một cây có khả năng và đôi khi bỏ lỡ thông tín.
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 15
Giá trị Bootstraping là một phương pháp giải quyết các bất định của bài toán thống kê
tính cho các node. Qua việc liên tục lấy mẫu các cây thông qua việc xáo trộn bộ dữ liệu đi một
chút, và giá trị được báo cáo là phần trăm lặp lại bootstrap mà node đó được chỉ ra. Thế nên
100% nghĩa là node đó xuất hiện ở tất cả các lần lặp lại bootstrap. Giá trị bootstrap cho thấy mức
độ thống nhất và ổn định của cây, nếu trên 95% thì được xem là có ý nghĩa thống kê.
3. Cây phát sinh chủng loại phát sinh trên toàn bộ genome
Phân tích phát sinh chủng loại (Phylogenetics) so sánh và phân tích các trình tự của một
gene duy nhất, hay một số ít các gene cũng như nhiều loại dữ liệu khác. Phylogenomics là sự
giao thao của lĩnh vực tiến hóa và genomic. Phylogenomics so sánh toàn bộ genome hay tối thiểu
là một phần lớn của nghiên cứu genome.
Bốn mảng lớn của phylogenetics là: dự đoán chức năng gene; thành lập và phân loại mối
quan hệ tiến hóa; tiến hóa của họ gene; và phán đoán sự di truyền ngang các gene.
Khó khăn của phylogenomics so với việc xây dựng cây chủng loại phát sinh là khối
lượng công việc lớn do phải so sánh toàn bộ các trình tự gene, có sự xuất hiện biến dị giữa các
gene hay khác biệt về lịch sử tiến hóa của gene.
So sánh genome (nhất là vi khuẩn) chỉ ra rằng trong tiến hóa một lượng lớn gene đã được
truyền ngang từ loài này qua loài khác. Điều này đã thay đổi đáng kể các đặc tính sinh thái, bệnh
hoc của các loài vi khuẩn. Truyền gene ngang tạo ra một genome năng động trong đó DNA
ngoại lại được đưa vào hay xóa mất khỏi nhiễm sắc thể. Đó là một yếu tố gây nhiễu tiềm tàng
trong việc xây dựng cây chủng loại phát sinh dựa vào trình tự của một gene.
Homologs là những gen hoặc protein tương tự nhau do có chung tổ tiên hoặc nguồn gốc chung của chúng.
Orthologs là hai gen tương tự nhau ở hai loài khác nhau có chung một tổ tiên
Paralog là hai gen trong cùng một bộ gen là sản phẩm của một sự kiện nhân đôi gen của gen ban đầu.
Gene Ancestral Histione H1 ban đầu khi nhân đôi đã tạo ra hai paralog là Histone H1.1
và Histone H1.2. Ở người và tinh tinh, đều có paralog này. Histone H1.1 ở người và Histone
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 16
H1.1 ở tinh tinh gọi là một ortholog. Tất cả các gene histone nhân lên từ gene Ancestral histone
H1 ban đầu gọi là Homologs. Ở vi khuẩn E.coli xuất hiện một gene tổng hợp nên chuỗi protein
với chức năng tương tự như histone H1.1 ở người, ta gọi đây là một analog.
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 17
Phần 4. DỰ ĐOÁN GENE
1. Dự đoán và chú giải gen
Genome người có kích thước khoảng 3,2 tỷ base tuy vậy chỉ có 1,5% trong số mang
coding mã cho mRNA, do đó còn rất nhiền các trình tự chưa xác định được vai trò. Việc dự đoán
và chú giải gene giúp cho ta hiểu rõ hơn về cấu trúc genome và cũng tìm hiểu về các quá trình
tạo sản phẩm từ gene, phát sinh bệnh lý,…
Với gene phân mảnh, các đoạn exon thường bị ngăn cách bởi các intron. Mỗi intron
thường bắt đầu bằng GT và kết thúc bằng AG, trước AG trong intron khoảng 20 base có một
trình tự ngắn là CTGAC gọi là điểm nhánh (branch point). Trong gene có các vị trí đặc biệt,
đóng vai trò cho quá trình phiên mã và nhân đôi. Promotor là vị trí khởi đầu phiên mã, với 70%
các promoter có trình tự TATA box. Vị trí kết thúc phiên mã là các vùng giàu GC theo sau là
vùng giàu AT, ngoài ra còn có vị trí có đuôi poly A (AATAAA). Codon mở đầu trên gene là
ATG (AUG trên RNA) và các codon kết thúc là UAA, UAG, UGA.
2. Phương pháp dự đoán và chú giải gen
Các phần mềm được sử dụng để dự đoán gene phổ biến là GENSCAN, Grail, MZEF,
FGeneH, Hexon, Genie,…tất cả đều sử dụng lập trình tự động để tìm ra giải pháp tối ưu.
Phương pháp sử dụng EST
là phương pháp được sử dụng phổ
biến. EST (expressed sequence
tag) là một phần trình tự ngắn của
cDNA, có thể được dùng để xác
định bản phiên mã, và là công cụ
phát hiện gene cũng như xác định
trình tự gene. Đặc điểm của EST
là có tính lặp lại cao, chất lượng
trình tự thấp, phản ảnh các gene
được thể hiện và có thể đặc trưng
cho từng mô trong từng giai đoạn.
Các bước để xác định gene bằng
EST đó là: (1) tách mRNA từ các
mô, tế bào (2) phiên mã cDNA
phản ánh bộ phận của RNAs (3)
tách dòng cDNA vào một vector
(chiều ngẫu nhiên) (4) giải trình tự gene.
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 18
Phần 5. MOTIF VÀ DOMAIN CỦA DNA VÀ PROTEIN 1. Motif và Domain
Motifs là trình tự các hình mẫu ngắn,
xuất hiện lặp lại và có liên quan tới chức năng
sinh học nhất định. Các phần mang motif giống
nhau nhiều khả năng có thể mang những chức
năng giống nhau, góp phần trong dự đoán chức
năng của sản phẩm từ trình tự.
Các mô típ trình tự cụ thể thường làm
trung gian cho một chức năng phổ biến, chẳng
hạn như liên kết protein hoặc nhắm mục tiêu đến
một vị trí dưới tế bào cụ thể, trong nhiều loại
protein. Do có trình tự giống nhau, nên có thể trong tế bào các motif sẽ đảm nhận làm tín hiệu
cho cơ chế tổng hợp và biểu hiện chức năng của các protein. Với các motif giống nhau nhưng ở
các protein khác nhau, có thể cùng đảm nhận những chức năng giống nhau từ đó tạo nên các họ
protein với cấu trúc và chức năng tương tự nhau. Dựa vào motif, người ta cũng có thể coi đây là
một phương pháp phân loại protein.
Domain (module) là các cấu trúc có thể thực hiện chức năng và tồn tại một cách độc lập
với phần còn lại của protein. Chúng thường hình thành các đơn vị chức năng. Domain bám vào
các phân tử khác trong tế bào, thực hiện nhiệm vụ là con đường dẫn truyền tín hiệu hay đóng vai
trò trong quá trình xúc tác các phản ứng hóa – sinh.
Hiện nay người ta xác định rõ rằng protein thực hiện các chức năng của chúng chủ yếu
thông qua các domain cấu thành chúng. Do đó các domain được coi là đơn vị mà qua đó protein
phát triển. Mặc dù các domain có thể có các chức năng riêng biệt khi được kiểm tra riêng lẻ, sự
kết hợp của chúng trong một protein nhất định là điều làm phát sinh vai trò tổng thể của nó trong
các quá trình tế bào. Như vậy, trước tiên chúng ta phải hiểu các domain riêng lẻ và sau đó điều
tra cách chúng đóng góp vào chức năng protein.
Trong thực nghiệm, người ta có thể tạo ra các protein bị mất đi một hay một vài domain,
sau đó tiến hành các thí nghiệm tương tự nhau để đưa ra nhận định về chức năng của domain với protein.
Ví dụ về motif và domain:
* C2H2 Zinc-finger là một trong những motif bám DNA phổ biến nhất của nhân thực, có mặt
trong nhân tố phiên mã TFIIIA
* TAZ Zinc-finger có mặt ở CBP acetyltranferase
* Helix-turn-helix được tìm thấy trong tất cả các protein bám DNA có chức năng điều hòa biểu hiện gene.
2. Các phương pháp tìm kiếm motif và domain (module)
Có 3 phương pháp chủ yếu để tìm kiếm motif và protein:
a. Phương pháp liệt kê
Phương pháp này sử dụng cách tiếp cận rộng nhất, xem xét tất cả các motifs có khả năng
và ít có hạn chế. Một số chương trình như WeederWed và YMF sử dụng phương pháp này để
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 19
tìm kiếm. Tuy vậy, với một nguồn dữ liệu tìm kiếm lớn thì phương pháp này tốn nhiều công sức và thời gian.
b. Phương pháp tối ưu hóa xác định
Phương pháp này xem xét một mô hình tối ưu hóa dự đoán (EM) và một ma trận trọng số
vị trí. EM tiến hành ước lượng lặp lại khả năng với dữ liệu hiệu có, dùng các thông số và quan
sát hiện tại để xây dựng lại cấu trúc sau đó dùng cấu trúc và quan sát để tái ước lượng các thông số.
MEME (Multiple EM for Motif Elicitation) là chương trình sử dụng thuật toán này.
Bắt đầu từ một vị trí, EM luân phiên giữa các vị trí chỉ định và mô hình motif đang cập nhật.
Thực hiện lặp lại một lần với mỗi n-mer ở trình tự đích, chọn motif tốt nhất từ vị trí này và sau
đó lặp lại cho đến khi motif đó không đổi nữa. Không gian tìm kiếm tăng đáng kể với sự tăng số
lượng trình tự và độ dài trình tự.
c. Phương pháp tối ưu hóa xác suất
Sử dụng cách tiếp cận của Gibbs, thực hiện một cách ngẫu nhiên mô hình tối ưu hóa dự
đoán (EM). Một n-mer từ mỗi trình tự được lấy ngẫu nhiên để xác định mô hình ban đầu. Trong
các lần lặp lại sau, một trình tự bị loại bỏ và mô hình được tính toán lại. Chọn vị trí mới của
motif trên trình tự lần lặp cho đến khi không đổi.
Các chương trình AlignAce, Gibbs Motif Sampler sử dụng thuật toán này. Chương trình
SMART là một chương trình phổ biến hơn cả. Nó cho phép xác định và giải thích vè các domain
vận động về mặt di truyền và phân tích các cấu trúc của domain. SMART dựa vào so sánh nhiều
trình tự của các thành viên, cơ sở dữ liệu với hơn 400 domains thuộc 54,000 protein. Chương
trình này tìm kiếm trong cơ sở dữ liệu bằng công cụ HMMs. SMART có hai chế độ: Normal
SMART thì cơ sở dữ liệu chứa Swiss-Prot, SP-TrEMBL và Ensembl proteomes; và chế độ
Genomic SMART chỉ có các proteasome đã được giải trình tự hoàn toàn mới được sử dụng.
Để xem xét một kết quả tìm kiếm motif và domain là hiệu quả hay không, ta cần quan
tâm đến các yếu tố về tính đặc trưng nhóm hay đặc trưng vị trí; đặc hiệu trình tự và tính đồng dạng của nó.
3. Protein – dự đoán cấu trúc và chức năng
Nghiên cứu cấu trúc chức năng của protein người ta quan tâm đến các yếu tố: bám màng
hay hòa tan, cấu trúc bậc 2, tính ưu nước – kỵ nước, biến đổi sau dịch mã, vị trí trong tế bào, các
motif và domain có trong protein,….
Bằng việc tìm hiểu các yếu tố này, người ta có thể dự đoán ra được chức năng của
protein. Với những protein mới, dựa trên những thông tin đã biết ở các protein tương đồng với
nó, người ta có thể dự đoán được chức năng. Xác định motifs trong protein giúp khẳng định như
những yếu bảo thủ trong tiến hóa và vai trò quan trọng của chúng. Việc các protein khác nhau
mang các cấu trúc và đặc tính, chức năng tương tự nhau có thể xác định mô hình, dấu hiệu để
nhận biết một họ protein.
Hiện nay, các cơ sở dữ liệu nguồn thường được các công cụ, servers liên quan sử dụng là
ExPASy Proteimics Server và Uniprot. Các chương trình được sử dụng phổ biến là PROSITE,
Pfam, BLOCKS, PSORT, TargetP, ProtFun,…
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC 20
Trong phân tử protein, ở cấu trúc bậc 3 hay bậc 4 các gốc kỵ nước thường ở phần “lõi”
giữa các phân tử, các gốc ưa nước ở trên bề mặt phân tử. Lực liên kết của các nhóm kỵ nước,
những nhóm không phân cực như CH2; CH3 trong valine, leucine, isoleucine,
phenylalanine…Nước trong tế bào đẩy các gốc này lại với nhau, giữa chúng xảy ra các lực hút
tương hỗ và tạo thành các đuôi kỵ nước trong phân tử protein. Do có cấu trúc bậc ba mà các
protein có được hình thù đặc trưng và phù hợp với chức năng của chúng. Ở các protein chức
năng như enzym và các kháng thể, protein của hệ thống đông máu…thông qua cấu trúc bậc ba
mà hình thành được các trung tâm hoạt dộng là nơi thực hiên các chức năng của protein.
ĐC TIN SINH HỌC – VŨ HẢI DƯƠNG – K64 SINH HỌC
Tài liệu liên quan:
-

Cách Tiếp Cận và Ứng Dụng trong Khoa Học | Tin sinh học | Trường Đại học Khoa học Tự nhiên, Đại học Quốc gia Hà Nội
60 30 -

Bài báo cáo Nghiên cứu về gen nosZ của Pseudomonas | Tin sinh học | Trường Đại học Khoa học Tự nhiên, Đại học Quốc gia Hà Nội
58 29 -

Hướng Dẫn Chuẩn Bị Báo Cáo Thí Nghiệm Bioinformatics | Tin sinh học | Trường Đại học Khoa học Tự nhiên, Đại học Quốc gia Hà Nội
75 38 -

Tài liệu ôn thi | Tin sinh học | Trường Đại học Khoa học Tự nhiên, Đại học Quốc gia Hà Nội
59 30 -

Bài Tập BLAST Trong Tin Sinh Học B2 - Tìm Kiếm Trình Tự Gen | Tin sinh học | Trường Đại học Khoa học Tự nhiên, Đại học Quốc gia Hà Nội
68 34