Sử dụng công cụ tin-sinh học giải trình tự thế hệ mới trong tìm các đột biến của ca bệnh Seckel
được sưu tầm và soạn thảo dưới dạng file PDF để gửi tới các bạn sinh viên cùng tham khảo, ôn tập đầy đủ kiến thức, chuẩn bị cho các buổi học thật tốt. Mời bạn đọc đón xem!
Môn: Hóa y 11 tài liệu
Trường: Đại học Y Dược Thành phố Hồ Chí Minh 379 tài liệu
Tác giả:





Preview text:
Nghiên cứu Y học
Y Học TP. Hồ Chí Minh * Tập 22 * Số 2 * 2018
SỬ DỤNG CÔNG CỤ TIN-SINH HỌC GIẢI TRÌNH TỰ
THẾ HỆ MỚI TRONG TÌM CÁC ĐỘT BIẾN CỦA CA BỆNH SECKEL
Huỳnh Kim Hiệu1, Bùi Chí Bảo2, Nguyễn Thị Huỳnh Nga3, Nguyễn Minh Hiệp3, Đào Trọng Thức4,
Võ Văn Thành Niệm2 TÓM TẮT
Giới thiệu: Giải trình tự thế hệ mới hay còn gọi bằng thuật ngữ tiếng Anh là Next Generation Sequencing
– NGS là một trong những đột phá về công nghệ sinh học phân tử cũng như tin sinh học và có rất nhiều ứng
dụng trong việc phát hiện các đột biến gây bệnh trên quy mô toàn bộ gen. Hội chứng Seckel là một bệnh hiếm
thuộc nhóm bệnh lùn cân đối (Primordial dwarfism) và việc sử dụng giải trình tự thế hệ mới, hay cụ thể ở đây là
giải trình tự exome (Whole Exome Sequencing – WES) để chẩn đoán, tìm hiểu về các gen có ảnh hưởng và di
truyền của bệnh là một hướng đi hiệu quả.
Mục tiêu: Tìm những đột biến trên exome có ảnh hưởng tới hình thành hội chứng Seckel và di truyền của
bệnh qua việc so sánh với exome người thân.
Phương pháp: Toàn bộ DNA lấy từ máu của bệnh nhân và người thân sẽ được chiết tách, giải trình tự sẽ
được thực hiện trên bộ exome và biến đổi sẽ được đánh giá và so sánh.
Kết quả: Trong nghiên cứu của chúng tôi, đột biến tại 15 gen liên quan được lọc ra từ dữ liệu NGS bằng
EXCEL theo phương án được nêu trên. Trong 23 đột biến tìm được, 22 đột biến đều đã có trên cơ sở dữ liệu,
chúng tôi tìm được một đột biến tạo bộ ba mã dừng (stopgain) là một đột biến mới chưa được công bố và sẽ cần
kiểm chứng lại bằng giải trình tự Sanger.
Kết luận: Kết quả đột biến từ NGS gợi ý một số hướng nghiên cứu tiếp tục cho ca bệnh trên; theo giả thiết
chúng tôi đưa ra, khả năng ca bệnh trên là MOPD loại II. Để khẳng định và tìm hiểu sâu hơn, chúng tôi dự kiến
sẽ giải trình tự Sanger cho các đột biến tiềm năng, đồng thời phân tích bộ gen của bố/mẹ bệnh nhân để khảo sát
đặc điểm di truyền của bệnh.
Từ khóa: Hội chứng Seckel, Hội chứng Meier-Gorlin, Giải trình tự thế hệ mới ABSTRACT
USING BIOINFORMATICS TOOLS AND WHOLE EXOME SEQUENCING FOR THE DETECTION OF
ACTIONABLE MUTATIONS IN SECKEL CASE REPORT
Huynh Kim Hieu, Bui Chi Bao, Nguyen Thi Huynh Nga, Nguyen Minh Hiep, Dao Trong Thuc,
Vo Van Thanh Niem * Y Hoc TP. Ho Chi Minh * Vol. 22 - No 2- 2018: 27 - 33
Background: Next generation sequencing or NGS is one of the breakthroughs in molecular biotechnology
and bioinformatics; it owns numerous applications in identifying causal genes on a genomic scale. Seckel
Syndrome is a rare disorder belonging to the group of Primordial Dwarfism; using next generation sequencing, or
particularly in this paper, whole exome sequencing to diagnose, study genes and their heredity.
Objectives: Identifying mutations in the patient’s exome for finding related genes and compare them to the
patient’s mother for studying the disease’s heredity.
1 Phòng Hợp tác Quốc tế, ĐH Y Dược Tp. HCM 2 Trung tâm Y sinh học phân tử, ĐH Y Dược Tp. HCM
3 Trường Đại học Đà Lạt
4 Trường Đại học Quốc tế - Đại học Quốc Gia TP. HCM
Tác giả liên lạc: TS.Huỳnh Kim Hiệu, ĐT: 090 118 9780
Email: huynhkimhieu@ump.edu.vn 28
Y Học TP. Hồ Chí Minh * Tập 22 * Số 2 * 2018 Nghiên cứu Y học
Methods: Genomic DNA of patient and relative are extracted, whole exome sequencing is conducted on
those samples and the resulted variants are assessed and compared.
Results: In our study, 15 genetic mutations were sorted out using EXCEL as explained. 22 out of 23
mutations found have been described on databases. We discovered a novel stop gain mutation and need verifying
using Sanger sequencing.
Conclusions: NGS mutational results suggest some research directions for the case examined. According to
our hypothesis, the case could be classified as MOPD type II. For further studies, we will Sanger sequence the
potential mutations, as well as analyzing the parenalgenomic DNA to survey the genetic characteristic of the disease.
Keywords: Seckel Syndrome, Meier-Gorlin Syndrome, Next Generation Sequencing ĐẶT VẤN ĐỀ
Nhóm chúng tôi đã tiếp cận được với một
Hội chứng Seckel – Seckel Syndrome (SCKS)
bệnh nhân được chẩn đoán mắc SCKS, tuy
là một bệnh rối loạn di truyền hiếm thuộc nhóm
nhiên, với kiểu hình và các đặc điểm lâm sàng
bệnh lùn tương đối – Primordial Dwarfism.
phức tạp để xác định và phân loại, chúng tôi
Bệnh lùn tương đối, ngoài SCKS, còn bao gồm
quyết định sử dụng NGS trên mẫu bệnh trên, cụ Microcephalic Osteodysplactic Primordial
thể hơn, chúng tôi sử dụng giải trình tự toàn bộ
Dwarfsims loại I-III (MOPD I-III) và hội chứng
Meier-Gorlin (MGS)(5,7). Những đặc điểm lâm
exome để tập trung vào những gen liên quan với
sàng điển hình của SCKS là dị tật đầu nhỏ
SCKS. Thêm vào đó, chúng tôi còn mở rộng ra
(thường thể tích não giảm còn 1/3 thể tích thông
đánh giá tất cả các gen có liên quan tới các hội
thường) và dị tật nội tạng (biến đổi cấu trúc
chứng khác của nhóm bệnh lùn tương đối theo
răng, hàm thụt và mũi nhô), mức độ nghiêm bảng dưới đây:
trọng của bệnh đối với bệnh nhân thay đổi rất
Bảng 1. Tổng quan các gen liên quan đến các nhóm
nhiều với các bệnh nhân(7). Một số gen được
bệnh thuộc hội chứng lùn tương đối(5,7) (Theo Geister
phân loại theo chức năng có liên hệ tới bệnh là:
KA và cs. (2015) và Khetarpal P và cs. (2016))
gen liên quan đến phản ứng với sự hư hỏng MOPD I/III MOPD II MGS SCKS
DNA – ATR, RBBP8; gen liên quan tới trung tử - RNU4ATAC, ORC1, ORC4, ATR, RBBP8, CPAP (CENPJ), CEP152 PCNT (5,7). U4ATAC ORC6, CDT1, CENPJ,
Giải trình tự thế hệ mới - Next Generation NIN, XRCC4, CENPE CDC6 CEP152
Sequencing (NGS) là một công nghệ mới áp
ĐỐI TƯỢNG – PHƯƠNG PHÁP NGHIÊN CỨU
dụng việc giải trình tự song song trên nhiều
Đối tương nghiên cứu
đoạn DNA cùng một lúc; công nghệ này ưu
Nghiên cứu được thực hiện trên một ca bệnh
việt hơn so với giải trình tự Sanger về mặt
công suất cũng như hiệu quả kinh tế
được chẩn đoán là Seckel tại Việt Nam. (3,4). Cụ
thể, việc sử dụng giải trình tự Sanger cho các
Phương pháp nghiên cứu
gen nhiều exon hoặc nhiều gen là cực kỳ tốn
Tách chiết gDNA từ máu
kém so với NGS. Tuy nhiên, đối với NGS, kết
Mẫu gDNA từ máu ngoại vi được tách chiết
quả giải trình tự là một ngân hàng dữ liệu
bởi bộ Il ustra-blood genomic Prep Mini Spin Kit
khổng lồ, vì vậy, cần phải có kiến thức về tin
sinh học cùng với máy tính có khả năng xử lý
(GE Healthcare). gDNA sau khi thu nhận được
dữ liệu mạnh để có thể phân tích kết quả giải bảo quản ở -20oC. trình tự.
Đánh giá chất lượng gDNA 29 Nghiên cứu Y học
Y Học TP. Hồ Chí Minh * Tập 22 * Số 2 * 2018
Theo quy trình của nhà sản xuất
Amplicon Library để chuẩn bị cho load trên
DropSense96 (Agilent) hoặc Qubit 2.0. Cho 2 µl
chip tải 3-8Gb mẫu chạy giải trình tự bằng
gDNA sẽ được nạp vào 96 giếng và đo 3 lần lặp
cách theo các hướng dẫn trên màng hình của
máy theo chương trình MiSeq v2, Illumina
lại với 2 tỷ lệ OD: A260/230 và A260/280) theo
HiSeq 4000. Sau khi tách gDNA sẽ nạp 1 µl hướng dẫn của máy.
chạy trên gel 1% agarose để đánh giá đứt gãy
Chuẩn bị thư viện Amplicon (TruSeq Custom và suy thoái DNA.
Amplicon Index Kit, Illumina)
Quy trình phân tích dữ liệu NGS
Các gDNA sẽ được cắt ngẫu nhiên để tạo ra
Kiểm tra chất lượng (Quality Control)
phân mảnh nhỏ (~500 đến 1.000 bp). Đây là quá
Kết quả đọc thô của hệ thống giải trình tự
trình chuẩn bị sự lai của hỗn hợp các mảnh nhỏ
(định dạng file FASTQ) sẽ được kiểm tra bằng
của từng mẫu với adapter và barcode. Sau quá
bằng phần mềm FASTQC(2). FASTQC sẽ đánh
trình lai diễn ra, các mẫu sẽ tiến hành PCR với 2
giá các tiêu chí chất lượng và cung cấp một bảng
mastermix thương mại TDP1 và PMM2 trong
thống kê đánh giá dưới định dạng file HTML.
index amplication plate (96 giếng). Sau phản
Một số chỉ tiêu FASTQC đánh giá là: thống kê sơ
ứng, thêm 25 µl 50 mM NaOH trước khi cho vào
bộ (Basic Statistics), Điểm chất lượng của từng
chu trình PCR. Chạy chương trình PCR bao gồm
base (Per base sequence quality), Tỷ lệ %GC trên
một bước biến tính nhiệt ở 95°C trong 3 phút;
một chuỗi đọc (Per sequence GC content), v.v.
tiếp theo là 25 chu kỳ của 95°C trong 30 giây,
Những mức chuẩn cũng như cách phân tích
62°C trong 30 giây, 72°C trong 60 giây; theo sau
bảng thông kê được trình bày rất trực quan và
là một phần mở rộng cuối cùng của 72°C trong 5
dễ hiểu trên trang chủ của FASTQC.
phút; kết thúc ổn định mẫu ở 10°C. Nếu không
tiến tới giai đoạn tiếp theo sau khi hoàn thành
Chuẩn bị trước phân tích (Preprocessing)
PCR, các mẫu có thể được bảo quản ở 2-8°C đến
Sau khi thực hiện kiểm tra chất lượng bằng hai ngày.
FASTQC, kết quả được khuyến nghị đưa qua
Tinh sạch sản phẩm PCR và nội cân bằng hạt
một số phần mềm nhằm sàng lọc lại các đoạn từ Bead-based
đọc thô. Chúng tôi sử dụng công cụ
Trimmomatic(2), quy trình sàng lọc căn bản là
Khi PCR hoàn tất, ly tâm mẫu ở 1.000 x g
cắt các đoạn bắt (adapter) ở đầu 3’ nếu có và
trong1 phút ở 20°C. Chuyển 1 µl mỗi phản
cắt những base chất lượng thấp ở cuối các
ứng PCR qua 1 giếng có chứa 4 µl nước để chuỗi đọc thô.
pha loãng các mẫu tỷ lệ 1/5. Pipette lên và
xuống để trộn và thêm 2 ml mẫu PCR pha
Bắt cặp trình tự đọc thô (Alignment)
loãng 2 ml đệm vi lỏng-gel. Con dấu các dải /
Sau khi được xử lý, các chuỗi trình tự đọc
tấm. Lắc tại 1.800 rpm trong ít nhất 30 giây và
thô sẽ được bắt cặp lại với một đoạn DNA
máy ly tâm ở 1.000 x g trong 30 giây. Theo
khung mẫu (reference genome). Chúng tôi sử
giao thức của nhà sản xuất quy trình lần lượt
dụng BWA(8), một phần mềm mã nguồn mở áp
là: Library Normalization Plate (LNP), dụng thuật toán Burrows-Wheeler
Library Normalization Additives 1 (LNA1),
Transformation. Thuật toán này cho phép sự bắt
Library Normalization Wash 1,2.3, Library
cặp có những đoạn không tương tự ở một mức
Normalization Storage Buffer 1 chạy hỗn hợp
độ nhất định giúp tìm ra những đột biến điểm
trên một gel thể lỏng để đánh giá xem việc
cũng như những đột biến thêm bớt đoạn.
chuẩn bị thư viện tốt không. Sau khi kiểm tra
sản phẩm PCR. Hỗn hợp mẫu library ban đầu
sẽ được trộn với nhau, đánh dấu Pooled 30
Y Học TP. Hồ Chí Minh * Tập 22 * Số 2 * 2018 Nghiên cứu Y học
Xử lý dữ liệu sau bắt cặp (Post-alignment
đột biến; VQSR bao gồm hai bước: (1) tạo ra dữ processing)
liệu chuẩn cho các đột biến từ các nguồn dữ liệu
Việc xử lý dữ liệu sau bắt cặp chúng tôi sử
có sẵn (dbSNP, 1000 Genome Project, HapMap
dụng bao gồm hai bước: loại bỏ các đoạn trùng
v.v) và (2) lọc dữ liệu của mẫu bằng dữ liệu
lặp (Remove read duplicates) được cung cấp bởi
chuẩn vừa tạo và gán nhãn đạt (PASS) cho các
công cụ Picard và chuẩn hóa điểm chất lượng
mẫu vượt qua được các tiêu chuẩn.
của base (Base quality score recalibration) được
Gán nhãn đột biến với các đặc tính sinh học
cung cấp bởi công cụ GATK(9). Về việc loại bỏ các
và lâm sàng (Variant annotation)
đoạn trùng lặp, khi bắt cặp với đoạn DNA
Chúng tôi áp dụng ANNOVAR(10) để gán
khuôn mẫu, sẽ có những đoạn đọc thô là kết quả
nhãn các đột biến tìm được trên mẫu bệnh. Một
nhiễu do quá trình PCR mang tới, vì vậy, sử
số nguồn dữ liệu chúng tôi sử dụng là RefSeq,
dụng thuật toán của Picard sẽ đánh dấu và loại
dbSNP, v.v và một số thuật toán tiên đoán ảnh
bỏ những tín hiệu nhiễu này. Đối với chuẩn hóa
hưởng của đột biến (SIFT, PolyPhen). Tùy vào
điểm chất lượng của các base, mỗi hệ thống giải
lựa chọn của người dùng, ANNOVAR có thể
trình tự sẽ có cách tính điểm riêng, vì vậy, các
gán nhãn các đột biến về tên gen, chức năng của
điểm số này lệ thuộc rất nhiều vào từng hệ
vùng đột biến (intron hoặc exon), ảnh hưởng của
thống, GATK cung cấp công cụ dùng để chuẩn
đột biến trên exon (synonymous, non-
hóa điểm số này để tăng độ tin cậy ở các bước
synonymous, stop-gain, v.v), biến đổi cấu trúc tiếp theo.
của chuỗi amino acid, ID của đột biến trên các
Gọi ra các đột biến (Variant calling)
database như dbSNP và các tiên đoán của các
Sau khi có kết quả của việc bắt cặp, chúng tôi
thuật toán như SIFT(6), PolyPhen(1), v.v về mức
sử dụng công cụ HaplotypeCaller của GATK(9)
độ ảnh hưởng của đột biến lên chức năng
để gọi ra những đột biến điểm hay đột biến
protein. ANNOVAR sẽ cho ra một tập tin dưới
thêm bớt đoạn. Công cụ này hoạt động dựa trên
định dạng tab-delimited text, tập tin trên có thể
nguyên tắc đánh dấu những vùng có khác biệt
mở bằng EXCEL để phân loại ra các đột biến
(active region), loại bỏ thông tin bắt cặp tại vùng được quan tâm.
đó là hoàn toàn thực hiện lại việc bắt cặp trên KẾT QUẢ
những vùng đó nhằm tăng độ chính xác của việc
Bảng thống kê sơ bộ về đoạn đọc thô
gọi ra những đột biến.
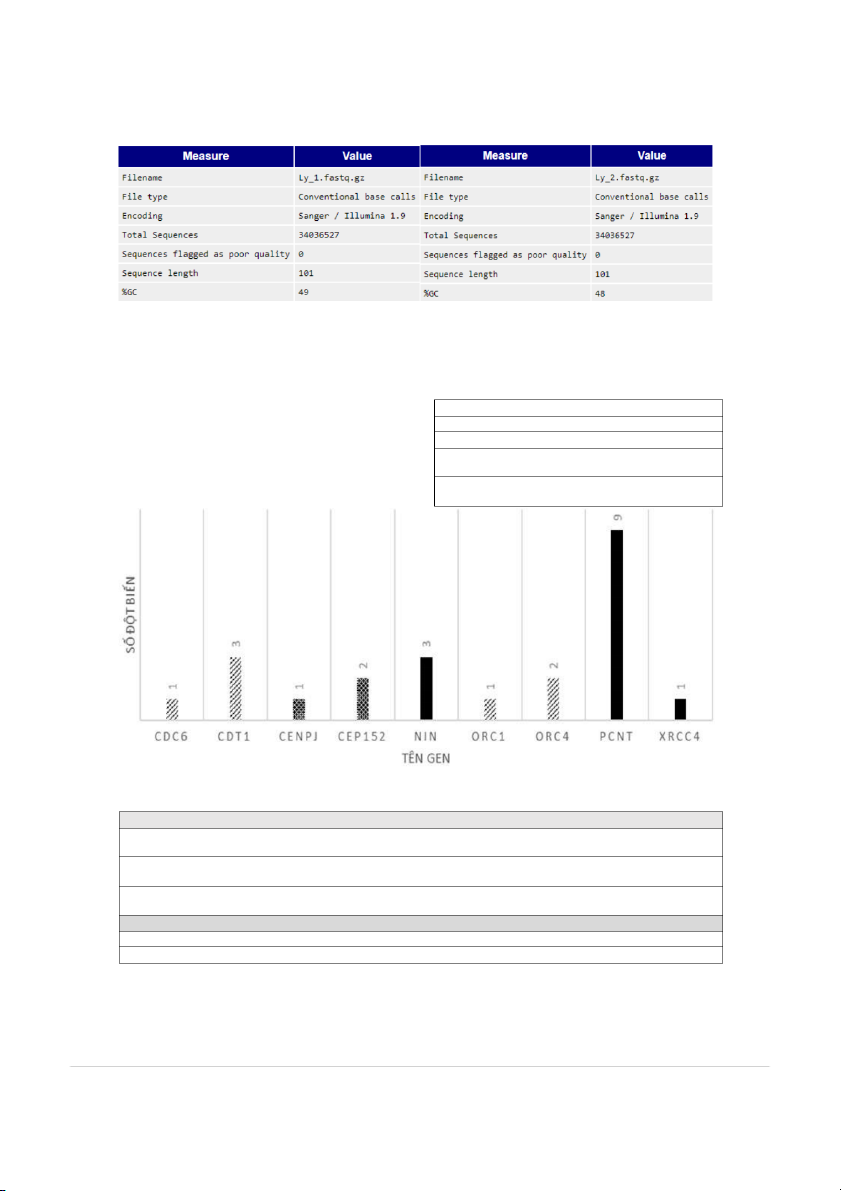
Kết quả thống kê cho mẫu bệnh, chúng tôi
Xử lý dữ liệu đột biến (Variant recalibration)
sử dụng giải trình tự hai đầu (paired-end), nên
Sau khi đột biến được gọi ra, thông thường,
với một mẫu bệnh, sẽ có hai tập tin thô.
những tiêu chuẩn phân loại về mặt chất lượng
Thông tin căn bản về hai đoạn đọc thô cho
bắt cặp sẽ được đặt ra và áp dụng để lọc dữ liệu
thấy, tổng số trình tự là 34036527 đoạn, với 0
đột biến. Tuy nhiên, việc sử dụng những tiêu
đoạn được đánh giá là chất lượng thấp, độ dài
chuẩn phân loại cứng ngắc (hard filters) như vậy
các đoạn là 101 và %GC rơi vào khoảng 49%.
là không tối ưu. GATK(9) cung cấp công cụ
Variant Quality Score Recalibration (VQSR) áp
dụng máy học (machine learning) để lọc lại các
Bảng 2. Thống kê cho hai đoạn đọc thô, forward (trái) và reverse (phải) 31 Nghiên cứu Y học
Y Học TP. Hồ Chí Minh * Tập 22 * Số 2 * 2018
Kết quả đột biến
Tổng hợp thông tin của tất cả các đột biến
Chúng tôi sử dụng EXCEL để lọc thủ công
tìm được phân loại theo nhóm bệnh được trình
các đột biến theo quy trình: (1) lọc đột biến theo bài trong các bảng 4,5,6:
những gen quan tâm (Bảng 1); (2) loại các đột
Bảng 4. Tổng hợp đột biến hai gen liên quan tới
biến tác động tới intron; (3) loại các đột biến SCKS
synonymous (không làm thay đổi axit amin). CENPJ
Chúng tôi tìm được một số đột biến không tác
CENPJ:NM_018451:exon2:c.A61T:p.M21L
động tới intron. Kết quả được thể hiện qua bảng CEP152 dưới đây:
CEP152:NM_001194998:exon20:c.C2740G:p.L914V,CEP1
52:NM_014985:exon20:c.C2740G:p.L914V
CEP152:NM_001194998:exon3:c.C161T:p.S54L,CEP152:
NM_014985:exon3:c.C161T:p.S54L
Hình 1: Tổng quan đột biến non-synonymous không tác động tới intron trên các gen liên quan.
Bảng 5. Tổng hợp đột biến trên các gen liên quan tới MOPD I-III NIN
NIN:NM_016350:exon27:c.C3661G:p.Q1221E,NIN:NM_020921:exon28: c.C5800G
:p.Q1934E,NIN:NM_182944:exon28:c. C5800G:p.Q1934E,NIN:NM_182946:exon28:c.C5800G:p.Q1934E
NIN:NM_020921:exon18:c.G3959A:p.G1320E,NIN:NM_182944:exon18:c.G3959A:p.G1320E,
NIN:NM_182946:exon18:c.G3959A:p.G1320E
NIN:NM_020921:exon18:c.A3374C:p.Q1125P,NIN:NM_182944:exon18:c.A3374C:p.Q1125P,
NIN:NM_182946:exon18:c.A3374C:p.Q1125P PCNT NM_006031:c.-105del-
PCNT:NM_001315529:exon10:c.C1262T:p.T421I,PCNT:NM_006031:exon10:c.C1616T:p.T539I 32
Y Học TP. Hồ Chí Minh * Tập 22 * Số 2 * 2018 Nghiên cứu Y học
PCNT:NM_001315529:exon13:c.G1757A:p.G586E,PCNT:NM_006031:exon13:c.G2111A:p.G704E
PCNT:NM_001315529:exon15:c.A2281G:p.T761A,PCNT:NM_006031:exon15:c.A2635G:p.T879A
PCNT:NM_001315529:exon15:c.T2759C:p.V920A,PCNT:NM_006031:exon15:c.T3113C:p.V1038A
PCNT:NM_001315529:exon26:c.A4561G:p.I1521V,PCNT:NM_006031:exon26:c.A4915G:p.I1639V
PCNT:NM_001315529:exon34:c.C7138T:p.Q2380X,PCNT:NM_006031:exon34:c.C7492T:p.Q2498X
PCNT:NM_001315529:exon37:c.G7623C:p.Q2541H,PCNT:NM_006031:exon37:c.G7977C:p.Q2659H
PCNT:NM_001315529:exon38:c.A8021G:p.Q2674R,PCNT:NM_006031:exon38:c.A8375G:p.Q2792R XRCC4
NM_001318012:exon8:c.894-1G>A;NM_022406:exon8:c.894-1G>A
Bảng 6. Tổng hợp các đột biến các gen liên quan tới MGS ORC1
ORC1:NM_001190818:exon9:c.C1397T:p.T466M,ORC1:NM_001190819:exon9:c.C1382T:p.T461M,ORC1:NM_004153:exon 9:c.C1397T:p.T466M ORC4
ORC4:NM_001190882:exon4:c.A11G:p.N4S,ORC4:NM_002552:exon5:c.A233G:p.N78S,ORC4:NM_181741:exon5:c.A233G:
p.N78S,ORC4:NM_181742:exon5:c.A233G:p.N78S,ORC4:NM_001190879:exon6:c.A233G:p.N78S
NM_002552:c.-44745T>C;NM_001190882:c.-47962T>C;NM_001190881:c.-61864T>C;NM_001190879:c.-
44745T>C;NM_181741:c.-44745T>C CDT1
CDT1:NM_030928:exon5:c.T700C:p.C234R
CDT1:NM_030928:exon5:c.A784G:p.T262A NM_030928:c.*92C>T CDC6
CDC6:NM_001254:exon10:c.G1321A:p.V441I BÀN LUẬN
chúng tôi sẽ tiến hành kiểm chứng lại bằng
Việc chẩn đoán các bệnh hiếm là rất phức
giải trình tự Sanger để đánh giá chính xác về
tạp và đòi hỏi sự kết hợp của nhiều phương đột biến này.
phướng tiếp cận. NGS là một trong những công
Về độ tin cậy của các đột biến (có hay
cụ có thể được sử dụng, trong nghiên cứu này,
không đột biến tồn tại tại các vị trí sau phân
chúng tôi đã chứng minh được sự hiểu quả của
tích), việc sử dụng công cụ VQSR của bộ công
NGS trong việc quan sát và đánh giá trên rất
cụ GATK đã loại bỏ đi những đột biến chất
nhiều gen, bao gồm gen liên quan tới bệnh cũng
lượng thấp và đã được chứng minh tính hiệu
như gen liên quan tới các nhóm bệnh tương tự.
quả(10). Về độ tin cậy của kiểu gen (đồng hợp
Tuy nhiên, đối với dữ liệu đột biến lớn mà
hay dị hợp), các tiêu chí cần được đánh giá là
NGS đã tìm ra, cần phải có một phương án
kiểu gen GT (Genotype), độ sâu của vị trí đọc
hiệu quả để lọc các đột biến quan tâm; cụ thể,
AD (Allele Depth) và DP (Depth of Coverage),
trong nghiên cứu của chúng tôi, đột biến tại 15
điểm tin cậy của kiểu tất cả các kiểu gen PL
gen liên quan được lọc ra từ dữ liệu NGS bằng
(“normalized” Phred-scale likelihood of
EXCEL theo phương án được nêu trên. Trong
possible genotypes) và điểm tin cậy của kiểu
23 đột biến tìm được, 22 đột biến đều đã có
gen GQ được chọn (Quality of the assigned
trên cơ sở dữ liệu bao gồm các gen CDC6,
genotype). Để đánh giá được các tiêu chí này,
CEP152, ORC4, ORC1, XRCC4, PCNT, NIN,
người phân tích cần có sự hiểu biết cả về các
CENPJ, CDT1 tương tự nghiên cứu liên quan
thống kê được sử dụng và cách đánh giá
đến các nhóm bệnh thuộc hội chứng lùn tương chúng.
đối(5,7). Hơn thế nữa, chúng tôi tìm được một KẾT LUẬN
đột biến tạo bộ ba mã dừng (stop-gain) là một
Tóm lại, đối với ca bệnh trên, có thể kết
đột biến mới chưa được công bố. Vì vậy,
luận rằng về mặt lâm sàng hội chứng SCKS là 33 Nghiên cứu Y học
Y Học TP. Hồ Chí Minh * Tập 22 * Số 2 * 2018
chưa đủ thông tin và cần được kiểm chứng. 4.
Fox AJ, et al. (2016). Next Generation Sequencing for the
Kết quả đột biến từ NGS gợi ý một số hướng
Detection of Actionable Mutations in Solid and Liquid
Tumors. Journal of Visualized Experiments, (115):1-11.
nghiên cứu tiếp tục cho ca bệnh trên; theo giả 5.
Geister KA and Camper SA. (2015). Advances in Skeletal
thiết chúng tôi đưa ra, khả năng ca bệnh trên
Dysplasia Genetics. Annual Review of Genomics and Human Genetics, 16(1): 199-227.
là MOPD loại II. Để khẳng định và tìm hiểu 6.
Henikoff S. (2003). SIFT: Predicting amino acid changes that
sâu hơn, chúng tôi dự kiến sẽ giải trình tự
affect protein function. Nucleic Acids Research, 31(13): 3812-
Sanger cho các đột biến tiềm năng, đồng thời 3814. 7.
Khetarpal P, Das S, Panigrahi I and Munshi A. (2016).
phân tích bộ gen của bố/mẹ bệnh nhân để
Primordial dwarfism: overview of clinical and genetic aspects.
khảo sát đặc điểm di truyền của bệnh.
Molecular Genetics and Genomics, 291(1): 1-15. 8.
Li H and Durbin R. (2010). Fast and accurate long-read
TÀI LIỆU THAM KHẢO alignment with Burrows-Wheeler transform. Bioinformatics 1. ,26(5):589-595.
Adzhubei I, Jordan DM and Sunyaev SR. (2013). Predicting
Functional Effect of Human Missense Mutations Using 9.
Van der Auwera GA, et al. (2002). GATK Best Practices.
Current Protocol in Bioinformatics, 11(1110):11.10.1-11.10.33.
PolyPhen-2. Current Protocols in Human Genetics, 76 (7.20): 1-
10. Wang K, Li M, Hakonarson H (2010). ANNOVAR: Functional 41.
annotation of genetic variants from high-throughput 2.
Bolger AM, Lohse M and Usadel B. (2014). Trimmomatic: A
flexible trimmer for Illumina sequence data. Bioinformatics,
sequencing data. Nucleic Acids Res,38(16): e164. 30(15): 2114-2120. 3.
Boycott KM, Vanstone MR, Bulman DE and MacKenzie AE.
Ngày nhận bài báo: 02/11/2017
(2013). Rare-disease genetics in the era of next-generation
sequencing: discovery to translation. Nature Reviews Genetics,
Ngày phản biện nhận xét bài báo: 03/11/2017 14(10): 681-691.
Ngày bài báo được đăng: 10/03/2018 34
Tài liệu liên quan:
-

Bài thu hoạch. Thực hành Hóa học
43 22 -

Đề Giữa Kỳ Hóa Sinh Y | Đại học Y Dược Thành phố Hồ Chí Minh
80 40 -

Bài Tập Ôn Tập Hóa Sinh Y | Đại học Y Dược Thành phố Hồ Chí Minh
106 53 -

Đề cuối kì hóa sinh y | Đại học Y Dược Thành phố Hồ Chí Minh
125 63 -

Trắc nghiệm Hóa sinh y | Đại học Y Dược Thành phố Hồ Chí Minh
79 40