Sử dụng dữ liệu geneonic trong tìm kiếm các đích tác động của chất kháng nấm ở vi nấm candida spp | Báo cáo sinh học phân tử
So sánh trình tự nhằm tìm kiếm, phân loại trình tự chuỗi, gen vẫn là một bài toán lớn của sinh tin học. Phổ biến hiện nay là so sánh đa trình tự, nhiều phương pháp được sử dụng và nhiều phần mềm đã được đưa ra nhằm giải quyết bài toán khi tập dữ liệu đầu vào lớn. Tài liệu giúp bạn tham khảo, ôn tập và đạt kết quả cao. Mời đọc đón xem!
Môn: Phương pháp và kỹ thuật cơ bản trong sinh họcphân tử 11 tài liệu
Trường: Đại học Y Dược Thành phố Hồ Chí Minh 379 tài liệu
Tác giả:


















Preview text:
lOMoAR cPSD| 45470709
Khoa Dược Bộ môn Vi sinh - Ký sinh
CHUYÊN ĐỀ HỌC PHẦN SINH HỌC PHÂN TỬ Tên chuyên đề:
SỬ DỤNG DỮ LIỆU GENEOMIC
TRONG TÌM KIẾM CÁC ĐÍCH TÁC
ĐỘNG CỦA CHẤT KHÁNG NẤM Ở
VI NẤM CANDIDA SPP Học viên
: Ninh Thị Hoa Hường
Nguyễn Thị Khánh Linh
Nguyễn Hồng Ngọc
Võ Nguyễn Mỹ Ngân Lớp
: Cao học Dược lý – Dược lâm sàng Niên khóa : 2022 – 2024
Thành phố Hồ Chí Minh, năm 2022 lOMoAR cPSD| 45470709 MỤC LỤC
DANH MỤC CHỮ VIẾT TẮT ........................................................................................................... 2
DANH MỤC HÌNH ẢNH .................................................................................................................. 3
DANH MỤC BẢNG .......................................................................................................................... 3
TÓM TẮT .......................................................................................................................................... 4
1. GIỚI THIỆU ................................................................................................................................ 4
2. NỘI DUNG .................................................................................................................................. 6
2.1. Phương pháp thu nhận dữ liệu geneomic .............................................................................. 6
2.2. Nguyên tắc hoạt động của các phần mềm được sử dụng để phân tích dữ liệu và dự ........... 9
đoán kết quả .................................................................................................................................. 9
2.2.1. BLAST (Basic Local Alignment Search Tool) ............................................................... 9
2.2.2. BioEdit .......................................................................................................................... 18
2.3. Các nghiên cứu liên quan về ứng dụng dữ liệu geneomic trong tìm kiếm đích tác động
chất kháng nấm ở Candida spp ................................................................................................... 19
2.3.1. So sánh geneomic cho phép xác định các đích tác động của thuốc kháng nấm
(Comparative geneomics allowed the identification of drug targets against human fungal
pathogenes) [15] ...................................................................................................................... 19
2.3.2. Ứng dụng thư viện gene đột biến để nghiên cứu chức năng bộ gene Candida
albicans (Application of the Mutant Libraries for Candida albicans Functional
Geneomics) [10] ...................................................................................................................... 24
2.3.3. Tính thiết yếu của việc sử dụng “machine-learning” trong dự đoán và sàng lọc
tương tác có hệ thống các thư viện hóa học để xác định các mục tiêu kháng nấm
(Leveraging machine learning essentiality predictions and chemogeneomic interactions to
identify antifungal targets) ...................................................................................................... 27
KẾT LUẬN ...................................................................................................................................... 35
TÀI LIỆU THAM KHẢO................................................................................................................. 35 1 lOMoAR cPSD| 45470709
DANH MỤC CHỮ VIẾT TẮT Từ viết tắt
Nội dung đầy đủ BLAST
Basic Local Alignment Search Tool CPR
Conditional Promoter Replacement GRACE
Genee Replacement And Conditional Expression PCR Polymerase Chain Reaction PDB Protein Database Bank C.albicans Candida albicans 2 lOMoAR cPSD| 45470709 DANH MỤC HÌNH ẢNH
Hình 2.1. Phương pháp so sánh geneomic .............................................................................. 5
Hình 2.2. Hai bước của phương pháp GRACE nghiên cứu trên C.albicans........................... 7
Hình 2.3. Giao diện truy cập BLAST (NCBI) ........................................................................ 13
Hình 2.4. Nhập chuỗi truy cập hoặc tải lên tệp có chứa chuỗi [10] ........................................ 14
Hình 2.5. Chọn cơ sỡ dữ liệu để tìm kiếm [10] ....................................................................... 14
Hình 2.6. Chọn thuật toán và các tham số của thuật toán để tìm kiếm [10] ........................... 15
Hình 2.7. Chạy chương trình BLAST [10] ............................................................................. 15
Hình 2.8. Phương pháp thiết lập danh sách từ truy vấn k-chữ cái [13]................................... 16
Hình 2.9. Phân tích phát sinh gen giữa các loại nấm gây bệnh ở người được thực hiện bằng
phân tích Bayesian [15] ........................................................................................................... 19
Hình 2.10. Cấu trúc ba chiều dự đoán của protein TRR1 và KRE2 thu được bằng mô hình
tương đồng [15] ....................................................................................................................... 21
Hình 2.11. Xây dựng mô hình machine learning để dự đoán tính cần thiết và thử nghiệm trên
bộ sưu tập GRACE ban đầu [25]. ............................................................................................ 22
Hình 2.12. Kiểm tra độ chính xác của mô hình dự đoán bộ dữ liệu GRACE v2 [25] ............ 22
Hình 2.13. NP-BTA nhắm vào glutaminyl-tRNA synthetase của C. Albicans [25] ............... 25
DANH MỤC BẢNG
Bảng 2.1. Các gen mục tiêu tiềm năng được chọn để phát triển thuốc kháng nấm mới ......... 10 3 lOMoAR cPSD| 45470709 TÓM TẮT
So sánh trình tự nhằm tìm kiếm, phân loại trình tự chuỗi, gen vẫn là một bài toán lớn của
sinh tin học. Phổ biến hiện nay là so sánh đa trình tự, nhiều phương pháp được sử dụng và nhiều
phần mềm đã được đưa ra nhằm giải quyết bài toán khi tập dữ liệu đầu vào lớn. Kiến thức về
gen và chức năng gen cho phép nghiên cứu các biện pháp phòng bệnh hiệu quả, thay đổi chiến
lược nghiên cứu thuốc và quy trình khám phá thuốc mới. Dựa trên các phương pháp tìm kiếm,
tổng hợp và hệ thống các tài liệu khoa học, các kết quả nghiên cứu liên quan gần đây, chuyên đề
đã làm rõ ba mục tiêu chính của đề tài “Sử dụng dữ liệu geneomic trong tìm kiếm các đích tác
động của chất kháng nấm ở vi nấm Candida spp’’. Phương pháp so sánh geneomics
(Comparative geneomics), phương pháp tiếp cận thay thế gen và biểu hiện có điều kiện (Genee
Replacement And Conditional Expression - GRACE) là hai phương pháp thu nhận dữ liệu
geneomics được các nhà khoa học sử dụng trong nhiều năm trở lại đây. Cùng với đó, các phần
mềm hỗ trợ phân tích dữ liệu gene và dự đoán kết quả cũng được nâng cấp thường xuyên như
Basic Local Alignment Search Tool (BLAST) và BioEdit. Tính thiết yếu của việc sử dụng
“machine-learning” trong dự đoán và sàng lọc tương tác có hệ thống các thư viện hóa học để
xác định các mục tiêu kháng nấm, ứng dụng thư viện gen đột biến để nghiên cứu chức năng bộ
gen Candida albicans và so sánh geneomic cho phép xác định các đích tác động của thuốc kháng
nấm … là một số nghiên cứu nổi bật về lĩnh vực trên chứng minh tầm quan trọng của geneomics
trong nghiên cứu phát triển thuốc mới. 1. GIỚI THIỆU
Candida (albicans và non-albicans) là nguyên nhân phổ biến gây nhiễm nấm xâm lấn
bệnh viện. Các loài nấm Candida là một phần của hệ sinh vật đường tiêu hóa bình thường ở
người. Tuy nhiên; việc dùng liệu pháp kháng sinh phổ rộng, phẫu thuật đường tiêu hóa hay giảm
bạch cầu trung tính là những yếu tố nguy cơ gây nhiễm nấm nghiêm trọng. Có hơn 200 loài
Candida trong tự nhiên nhưng có khoảng trên 30 loài trong số chúng thực sự gây nhiễm ở người,
phổ biến nhất là C. albicans, C. glabrata, C. krusei, C. parapsilosis và C. tropicalis. Các loài
Candida khác nhau về tính nhạy cảm với các chất kháng nấm khác nhau [1]. 4 lOMoAR cPSD| 45470709
Nhiễm nấm xâm lấn thường gặp trên các bệnh nhân nặng, tỷ lệ tử vong cao, đặc biệt ở
những bệnh nhân suy giảm miễn dịch và các bệnh nhân ở khoa hồi sức tích cực. Tần suất nhiễm
nấm xâm lấn đang tăng lên nhanh chóng trong vòng hơn 30 năm qua (1976 – 1996) do tình trạng
sử dụng kháng sinh phổ rộng kéo dài, liệu pháp ức chế miễn dịch hoặc suy giảm miễn dịch mắc
phải, ung thư, sử dụng các thiết bị xâm lấn, dinh dưỡng qua đường tĩnh mạch… [2, 3]. Một trong
những vấn đề cấp bách hiện nay là một số loài Candida ngày càng đề kháng với các thuốc kháng
nấm hiện có. Điều đó đặt ra thách thức tìm kiếm các đích tác động mới từ đó khám phá và tổng
hợp ra các thuốc kháng nấm mới với hiệu quả điều trị cao và hạn chế tối thiểu các tác dụng không mong muốn.
Ngày nay, khoa học kỹ thuật hiện đại ngày càng phát triển và có những bước tiến vượt
bậc, đặc biệt trong lĩnh vực y dược học với việc giải mã trình tự ADN hay toàn bộ gene các vi
sinh vật hoặc con người. Sự ra đời của khái niệm hệ gene học (geneomic) là khoa học nghiên
cứu toàn bộ các gene của geneome trong cơ thể. Geneomic đã phát triển nhanh chóng trong hơn
một thập kỷ gần đây nhờ việc ứng dụng công nghệ giải trình tự gene kết hợp với tin sinh học
(bioinformatics), tập trung nghiên cứu các đích tác động và trị liệu mới, góp phần làm sáng tỏ
các nhóm gene quyết định đến hiệu quả, độc tính hoặc đề kháng thuốc.
Trên cơ sở đó, nhóm thực hiện chuyên đề “Sử dụng dữ liệu geneomic trong tìm kiếm các
đích tác động của chất kháng nấm ở vi nấm Candida spp’’ với ba mục tiêu sau:
Mục tiêu 1: Trình bày các phương pháp thu nhận dữ liệu geneomic.
Mục tiêu 2: Trình bày nguyên tắc hoạt động của các phần mềm được sử dụng để phân tích
dữ liệu và dự đoán kết quả.
Mục tiêu 3: Trình bày các nghiên cứu liên quan về ứng dụng dữ liệu geneomic trong tìm
kiếm đích tác động chất kháng nấm ở vi nấm Candida spp. 5 lOMoAR cPSD| 45470709 2. NỘI DUNG
2.1. Phương pháp thu nhận dữ liệu geneomic
2.1.1. Phương pháp so sánh geneomics (Comparative geneomics) 2.1.1.1 Định nghĩa
So sánh geneomics là một lĩnh vực nghiên cứu sinh học, trong đó các nhà nghiên cứu sử
dụng nhiều công cụ khác nhau để so sánh trình tự bộ gen hoàn chỉnh của các loài khác nhau.
Bằng cách so sánh cẩn thận các đặc điểm xác định các sinh vật khác nhau, các nhà nghiên cứu
có thể xác định chính xác các vùng tương đồng và khác biệt [4].
Phương pháp bao gồm so sánh số lượng gen, hàm lượng và vị trí gen, độ dài và số lượng
vùng mã hóa (được gọi là exon) trong gen, lượng DNA không mã hóa trong mỗi bộ gen và các
vùng được bảo tồn được duy trì ở cả nhóm sinh vật nhân sơ và sinh vật nhân thực.
Hình 2.1. Phương pháp so sánh geneomic [5] 2.1.1.2 Lợi ích
Việc xác định các trình tự DNA đã bảo tồn ở nhiều sinh vật khác nhau qua hàng triệu năm
- là một bước quan trọng để hiểu chính bộ gen. Nó xác định chính xác các gen cần thiết cho sự
sống và làm nổi bật các tín hiệu gene kiểm soát chức năng gen ở nhiều loài. Giúp chúng ta hiểu 6 lOMoAR cPSD| 45470709
thêm về những gen liên quan đến các hệ thống sinh học khác nhau, từ đó có thể chuyển thành
các phương pháp sáng tạo để điều trị bệnh và cải thiện sức khỏe con người.
So sánh geneomics cũng cung cấp một công cụ mạnh mẽ để nghiên cứu sự tiến hóa. Bằng cách
tận dụng và phân tích mối quan hệ tiến hóa giữa các loài và sự khác biệt tương ứng trong DNA
của chúng, các nhà khoa học có thể hiểu rõ hơn về cách thức hình dáng, hành vi và sinh học của
các sinh vật sống đã thay đổi theo thời gian.
Khi công nghệ giải trình tự DNA trở nên mạnh mẽ hơn và ít tốn kém chi phí hơn, so sánh
geneomics mang lại ứng dụng rộng rãi hơn trong nông nghiệp, công nghệ sinh học và động vật
học. 2.1.1.3 Ứng dụng
So sánh geneomics có ứng dụng rộng rãi trong lĩnh vực y học phân tử và tiến hóa phân tử.
Ứng dụng quan trọng nhất của bộ gen so sánh trong y học phân tử là xác định các mục tiêu thuốc
của nhiều bệnh truyền nhiễm.
Chi nấm Candida là một loại nấm lưỡng bội, có bộ gen được giải trình tự vào năm 2004.
Bộ gen được cung cấp để sàng lọc trên diện rộng các mục tiêu tiềm năng bao gồm tất cả các loại
gen chức năng và được phân loại thành các nhóm khác nhau như enzym, chất vận chuyển, thụ
thể, yếu tố phiên mã, v.v. Bộ gen là công cụ hữu ích để xác định các mục tiêu thuốc mới tiềm
năng, chẳng hạn như các gen thiết yếu và / hoặc những gen ảnh hưởng đến khả năng tồn tại của
tế bào được bảo tồn trong các sinh vật gây bệnh. Các phân tích so sánh về bộ gen của nấm đã
dẫn đến việc xác định nhiều mục tiêu giả định cho thuốc kháng nấm mới [6]. Khám phá này có
thể hỗ trợ thiết kế thuốc dựa trên mục tiêu để chữa bệnh nấm ở người. Nghiên cứu “Comparative
geneomics allowed the identification of drug targets against human fungal pathogenes” của
Abadio và cộng sự (2011) đã xác định các mục tiêu thuốc tiềm năng được áp dụng cho bệnh
nấm ở người sử dụng phương pháp hệ gen so sánh.
2.1.2. Phương pháp tiếp cận thay thế gene và biểu hiện có điều kiện (Genee Replacement
And Conditional Expression - GRACE)
Phương pháp tiếp cận thay thế gene và biểu hiện có điều kiện được sử dụng để đánh giá
tính thiết yếu của gen thông qua sự kết hợp giữa thay thế gen và biểu hiện gen có điều kiện. 7 lOMoAR cPSD| 45470709
Phương pháp GRACE bao gồm hai thao tác liên tiếp: (i) thay thế gen chính xác của một
alen và (ii) biểu hiện có thể kiểm soát được của alen còn lại bằng cách thay thế gen khởi đầu tự
nhiên bằng gen khởi động tetracycline (Tet) có thể điều chỉnh chặt chẽ.
Trong một ứng dụng của phương pháp này, Terry Roemer và cộng sự (2003) đã đánh giá
1152 gene của C. albicans bằng phương pháp GRACE, trong đó 567 gene được chứng minh
bằng thực nghiệm là cần thiết cho sự tăng trưởng – gen thiết yếu của C. albicans. Việc xây dựng
bộ sưu tập chủng đột biến có điều kiện này tạo điều kiện cho việc kiểm tra quy mô lớn các kiểu
hình cuối cùng của các gen thiết yếu. Thông tin này cho phép các mục tiêu thuốc ưu tiên được
chọn từ gen thiết yếu của C. albicans được thiết lập bởi thông tin kiểu hình có nguồn gốc từ cả
in vitro, chẳng hạn như kiểu hình diệt khuẩn so với kiểu hình đầu cuối tĩnh, cũng như in vivo
thông qua nghiên cứu độc lực bằng cách sử dụng các chủng có điều kiện trong mô hình lây
nhiễm ở động vật. Ngoài ra, sự kết hợp giữa phân tích kiểu hình và tin sinh học giúp cải thiện
hơn nữa việc lựa chọn mục tiêu thuốc từ bộ gen thiết yếu của C. albicans và các chủng đột biến
có điều kiện tương ứng của chúng có thể được sử dụng trực tiếp làm xét nghiệm toàn tế bào nhạy
cảm để sàng lọc thuốc.
Hình 2.2. Hai bước của phương pháp GRACE nghiên cứu trên C. albicans [7] Chú thích 8 lOMoAR cPSD| 45470709
Bước 1: Các chủng dị hợp tử được tạo ra bằng cách chuyển đổi chủng ban đầu của Candida
albicans (chủng thuần) - CaSS1 bằng cách sử dụng “PCR-geneerated disruption cassette” có
chứa điểm đánh dấu chọn lọc HIS3 được gắn với trình tự tương đồng thích hợp để thay thế chính
xác một alen của gene đích. Hai mã vạch riêng biệt (BC1, "up tag" và BC2, "up down") đã được
đưa vào cassette gián đoạn trong quá trình khuếch đại PCR. Hai cặp mồi kết hợp với các nhánh
chung (tương ứng là màu vàng và đỏ) bên cạnh mỗi " up tag" và " up down", cho phép khuếch
đại PCR đơn giảm của mã vạch nhận dạng biến dạng. Do đó, tất cả các chủng dị hợp tử đều
được gắn duy nhất với mã vạch nhận dạng chủng riêng biệt.
Bước 2: Các chủng dị hợp tử có mã vạch được biến nạp bằng cách sử dụng một cassette
thay thế promoter tetracycline do PCR tạo ra có chứa đánh dấu chọn lọc ưu thế SAT-1 được thiết
kế để biểu hiện trong C. albicans. Trình tự khung tương đồng được thêm vào trong quá trình
khuếch đại PCR để thay thế chính xác trình tự khởi đầu nội sinh của alen chủng thuần còn lại
bằng trình tự thay thế trình tự promoter Tet sau khi biến nạp.
2.2. Nguyên tắc hoạt động của các phần mềm được sử dụng để phân tích dữ liệu và dự đoán kết quả
2.2.1. BLAST (Basic Local Alignment Search Tool)
BLAST (Basic Local Alignment Search Tool) là công cụ được sử dụng rộng rãi để tính
toán sự tương đồng giữa trình tự nucleotide hoặc protein từ các sinh vật. Phần mềm hoạt động
dựa trên nguyên tắc so sánh AND với các trình tự cơ sở dữ liệu protein có sẵn và tính toán mức
độ trùng khớp có ý nghĩa thống kê. BLAST có thể được sử dụng để xác định các gene mới trong
bộ gene, tìm kiếm chức năng của các gene được giải trình tự, từ đó dự đoán mối quan hệ chức
năng và tiến hóa [8]. Có nhiều biến thể khác nhau của BLAST để sử dụng cho việc so sánh các
trình tự khác nhau [9,10]:
• MegaBLAST dùng để tìm kiếm nucleotide-nucleotide, được tối ưu hóa cho các trình tự
rất giống nhau (trong cùng một hoặc trong các loài có quan hệ họ hàng gần). Đầu tiên, phần
mềm sẽ tìm kiếm sự trùng khớp chính xác của 28 cơ sở, sau đó cố gắng mở rộng cơ sở ban đầu
đó khớp thành một căn chỉnh đầy đủ). 9 lOMoAR cPSD| 45470709
• BLASTN dùng để tìm kiếm các trình tự nucleotide-nucleotide xa hơn.
• BLASTP thực hiện so sánh trình tự protein-protein và thuật toán của nó là cơ sở của
nhiều thuật toán khác các loại tìm kiếm BLAST như BLASTX và TBLASTN.
• BLASTX tìm kiếm truy vấn nucleotide dựa trên cơ sở dữ liệu protein, dịch truy vấn một cách nhanh chóng.
• TBLASTN tìm kiếm một truy vấn protein dựa trên cơ sở dữ liệu nucleotide, dịch cơ sở
dữ liệu một cách nhanh chóng.
• PSI-BLAST trước tiên thực hiện tìm kiếm BLASTP để thu thập thông tin mà sau đó PSI-
BLAST sử dụng để tạo ra Ma trận chấm điểm cụ thể theo vị trí (PSSM). PSSM cho truy vấn có
độ dài N là ma trận N x 20. Mỗi cột trong dãy N tương ứng với một chữ cái trong truy vấn và
mỗi cột chứa 20 hàng. Mỗi hàng tương ứng với một dư lượng cụ thể và mô tả xác suất của các
chuỗi liên quan có dư lượng đó ở vị trí đó. Tiếp theo, PSI-BLAST có thể tìm kiếm cơ sở dữ liệu
về trình tự protein với PSSM này.
• RPSBLAST (BLAST theo vị trí cụ thể đảo ngược) có thể tìm kiếm rất nhanh một truy
vấn protein dựa trên cơ sở dữ liệu của PSSM thường được sản xuất bởi PSI-BLAST.
• DELTA-BLAST tạo PSSM với tìm kiếm nhanh RPSBLAST của truy vấn, sau khi tìm
kiếm PSSM này dựa trên cơ sở dữ liệu về trình tự protein.
Phiên bản BLAST đầu tiên đã được NCBI sản xuất vào khoảng năm 1990, với tính năng
chỉ thực hiện các bắt cặp không có khoảng trống nhưng cung cấp giá trị p cho phép người dùng
đánh kết quả có ý nghĩa thống kê hay không. Sau đó, PSI-BLAST ra đời dựa trên sự sửa đổi của
BLAST vào năm 1997, có thể tạo ra một PSSM và tìm kiếm cơ sở dữ liệu với nó. Cả hai phiên
bản BLAST này đều sử dụng bộ ngôn ngữ lập trình C của NCBI. Vào cuối năm 2009, NCBI bắt
đầu hỗ trợ phiên bản BLAST mới hơn (được gọi là BLAST +) dựa trên bộ công cụ C++ làm nền
tảng phát triển. Trang web NCBI BLAST được xây dựng bằng bộ ngôn ngữ C++ và BLAST+
[8, 10]. Để cải thiện hơn nữa hiệu suất của PSI-BLAST đối với việc phát hiện tương đồng protein
từ xa, một nghiên cứu được giám sát khung tìm kiếm hai lớp dựa trên PSI-BLAST (S2L-
PSIBLAST) được đề xuất. S2L-PSIBLAST bao gồm tìm kiếm hai cấp: tìm kiếm cấp một cung
cấp kết quả tìm kiếm chất lượng cao bằng cách sử dụng khung SMIBLAST và chiến lược liên 10 lOMoAR cPSD| 45470709
kết kép để lọc các chuỗi protein không tương đồng, tìm kiếm cấp hai phát hiện nhiều protein
tương đồng hơn bởi sự tương đồng của liên kết hồ sơ và danh sách xếp hạng chính xác hơn cho
các chuỗi protein được phát hiện đó có được bằng việc học cách xếp hạng chiến lược. Kết quả
thử nghiệm trên phiên bản cập nhật phân loại cấu trúc của protein-bộ dữ liệu điểm chuẩn mở
rộng cho thấy rằng S2L-PSIBLAST không chỉ cải thiện rõ ràng hiệu suất của PSI-BLAST mà
còn đạt được hiệu suất tốt hơn trên hai phiên bản cải tiến của PSI-BLAST là DELTA-BLAST và PSI-BLASTexB [9].
2.2.1.1. Cách sử dụng BLAST
Truy cập đường link https://blast.ncbi.nlm.nih.gov với giao diện sau:
Hình 1.3. Giao diện truy cập BLAST (NCBI)
Các bước thực hiện
• Bước 1: Lựa chọn chương trình
Người dùng lựa chọn một trong những chương trình từ database: BLASTp, BLASTn, BLASTx, tBLASTn, tBLASTx.
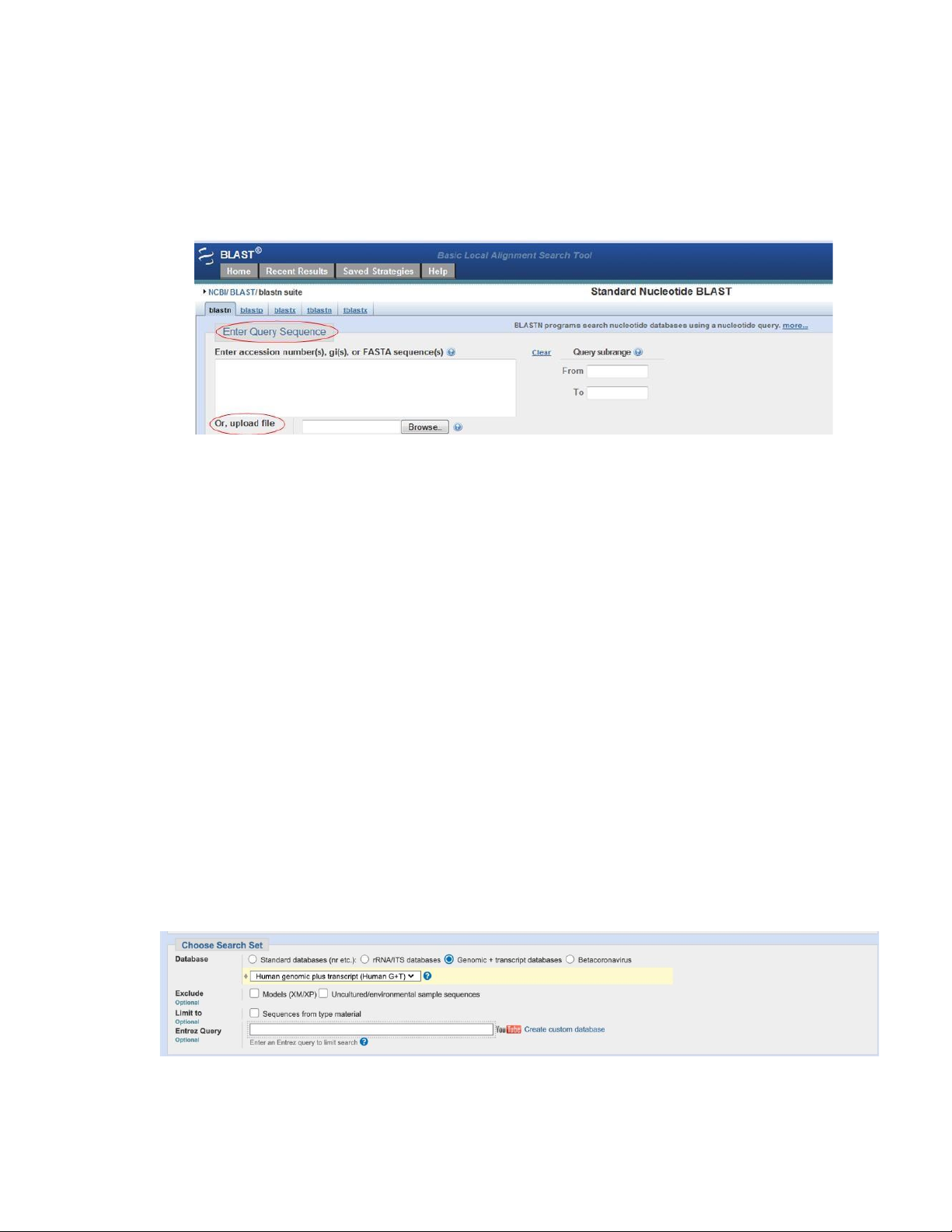
• Bước 2: Nhập chuỗi truy vấn hoặc tải lên tệp có chứa chuỗi 11 lOMoAR cPSD| 45470709
Nhập chuỗi truy vấn bằng cách dán chuỗi vào ô hoặc tải lên tệp FASTA có chuỗi để tìm
kiếm. Bước này tương tự đối với tất cả các chương trình BLAST. Người dùng có thể dùng mã
truy cập accession number hoặc GI hoặc truy cập vào FASTA để lấy trình tự.
Hình 2.4. Nhập chuỗi truy vấn hoặc tải lên tệp có chứa chuỗi [11]
• Bước 3: Chọn cơ sở dữ liệu để tìm kiếm
Người dùng trước tiên phải biết tất cả các cơ sở dữ liệu có sẵn là gì và loại trình tự nào
hiện diện trong các cơ sở dữ liệu đó. Ở bước này, tìm kiếm sự giống nhau về trình tự liên quan
đến việc tìm kiếm các trình tự tương tự của trình tự truy vấn từ các cơ sở dữ liệu đã chọn.
Chọn một trong các cơ sở dữ liệu sau:
- Standard databases: dữ liệu chuẩn, ví dụ: dữ liệu nucleotid (The nucleotide collection …)
- rRNA/ITS databases: dự liệu ARN ribosome.
- Geneomic + transcript databases (Human geneomic plus transcript, Mouse geneomic
plus transcript): dữ liệu trình tự bộ gene người + trình tự gene người được phiên mã và dữ liệu
trình tự bộ gene chuột + trình tự gene chuột được phiên mã.
- Betacoronavirus: dữ liệu về vi rút corona.
Hình 2.5. Chọn cơ sở dữ liệu để tìm kiếm [11] 12 lOMoAR cPSD| 45470709
• Bước 4: Chọn thuật toán và các tham số của thuật toán để tìm kiếm
Có các thuật toán khác nhau cho một số chương trình BLAST. Người dùng phải chỉ định
thuật toán cho chương trình BLAST. Nucleotide BLAST sử dụng các thuật toán như
MegaBLAST tìm kiếm các trình tự tương tự cao, MegaBLAST không liên tục tìm kiếm các trình
tự khác nhau nhiều hơn và BLASTn tìm kiếm các trình tự tương tự. Trong khi đó, đối với các
thuật toán BLAST protein như BLASTp, tìm kiếm sự tương đồng giữa protein truy Pvấn và cơ
sở dữ liệu protein, PSI-BLAST thực hiện lặp đi lặp lại tìm kiếm vị trí cụ thể, PHIBLAST tìm
kiếm một mẫu cụ thể (người dùng phải nhập mẫu để tìm kiếm trong hộp mẫu HI được cung cấp)
có trong trình tự so với các trình tự trong cơ sở dữ liệu, DELTA-BLAST là giúp tăng thời gian truy vấn BLAST.
Hình 2.6. Chọn thuật toán và các tham số của thuật toán để tìm kiếm [11]
Các tham số trong thuật toán xác định độ nhạy tìm kiếm.
“Max target sequences” (các trình tự mục tiêu tối đa): đặt các trình tự cơ sở dữ liệu ban
đầu tối đa phù hợp với BLAST tiết kiệm cho một truy vấn nhất định.
“Short queries” (các truy vấn ngắn): đã được kiểm tra cho phép BLAST tự động tối ưu
hóa cài đặt cho các truy vấn 30 cơ sở/dư lượng hoặc ngắn hơn.
“Expect threshold” (ngưỡng kỳ vọng): lọc ra các kết quả phù hợp ít quan trọng hơn, với
giá trị kỳ vọng trên cài đặt.
“Word size” (kích thước từ): cho phép xác định độ dài của chuỗi (Mục 2.1.3) với độ dài
càng nhỏ thì càng tăng tính nhạy cảm của kết quả.
“Max matches in a query range” (kết quả khớp tối đa trong một phạm vi truy vấn):
giới hạn các kết quả khớp được lưu vào một vùng nhất định của truy vấn (chẳng hạn như từ lặp 13 lOMoAR cPSD| 45470709
lại) để có thể báo cáo kết quả khớp với vùng khác của truy vấn. Cài đặt mặc định là “0” có
nghĩa là không có giới hạn.
• Bước 5: Chạy chương trình BLAST
Nhấn vào nút Blast ở cuối trang để chạy chương trình [11].
Hình 2.7. Chạy chương trình BLAST [11]
2.2.1.2. Nguyên tắc hoạt động của BLAST
BLAST thường có các Cặp phân đoạn điểm cao (High-scoring Segment Pairs – HSP) nằm
trong một liên kết có ý nghĩa thống kê (Mục 2.2.1.3 và 2.2.1.4). Nguyên tắc hoạt động chính của
phần mềm là tìm kiếm các HSP giữa chuỗi truy vấn và các chuỗi hiện có trong cơ sở dữ liệu, sử
dụng phương pháp heuristic gần đúng với thuật toán Smith-Waterman. Tuy nhiên, cách tiếp cận
toàn diện của Smith-Waterman quá chậm để tìm kiếm cơ sở dữ liệu bộ gene lớn như GeneBank.
Do đó, thuật toán BLAST sử dụng phương pháp heuristic kém chính xác hơn thuật toán Smith-
Waterman nhưng nhanh hơn 50 lần [12]. Tốc độ và độ chính xác tương đối tốt của BLAST là
một trong những cải tiến kỹ thuật quan trọng của các chương trình BLAST. 14 lOMoAR cPSD| 45470709
Để chạy phần mềm, BLAST yêu cầu một chuỗi truy vấn (chuỗi mẫu) để tìm kiếm và một
chuỗi để tìm kiếm (còn gọi là chuỗi đích) hoặc một cơ sở dữ liệu trình tự chứa nhiều chuỗi1.
Trình tự “truy vấn” BLAST được cung cấp dưới dạng các chuỗi ký tự của mã nucleotide hoặc
axit amin đơn dòng bắt đầu bằng ký hiệu “>” và chứa các số nhận dạng và thông tin mô tả. Định
dạng này được gọi là FASTA. Cơ sở dữ liệu BLAST được xây dựng từ các trình tự được định
dạng FASTA được nối với nhau bằng cách sử dụng một chương trình có tên “formatdb” tạo ra
hỗn hợp các tệp được mã hóa nhị phân và ASCII (American Standard Code for Information
Interchange - Chuẩn mã trao đổi thông tin Hoa Kỳ) chứa các trình tự và thông tin chỉ mục được
sử dụng trong quá trình tìm kiếm BLAST [11, 13].
BLAST tìm kiếm bằng cách đánh dấu tất cả các ký tự có độ dài nhất định trong theo vị trí
bắt đầu của chúng trong trình tự truy vấn. Người dùng có thể xác định độ dài của chuỗi,
được gọi là “word size”. Phạm vi cho phép đối với "word size" thay đổi tùy theo chương trình
BLAST được sử dụng; các giá trị điển hình là 3 cho các tìm kiếm trình tự protein-protein và 11
cho các tìm kiếm nucleotide thành nucleotide. Sau đó, BLAST sẽ quét cơ sở dữ liệu để tìm kiếm
các kết quả phù hợp giữa các “từ” trong trình tự truy vấn và các chuỗi được tìm thấy trong chuỗi
cơ sở dữ liệu. Đối với tìm kiếm protein, điểm được xác định bằng cách sử dụng ma trận thay
thế, điểm này phải vượt quá một ngưỡng quy định. Khi tìm thấy một từ phù hợp, BLAST sẽ mở
rộng cả về phía trước và phía sau từ đối chiếu để tạo ra một sự liên kết trong đối chiếu protein.
BLAST sẽ tiếp tục phần mở rộng này miễn là điểm bắt cặp tiếp tục tăng hoặc cho đến khi nó
giảm xuống điểm số âm gây ra do điểm không khớp [11].
Tổng quan về thuật toán BLAST như sau [14]:
• Loại bỏ vùng có độ phức tạp thấp hoặc các vùng lặp lại trình tự trong chuỗi truy vấn
"Vùng có độ phức tạp thấp" có nghĩa là vùng của một chuỗi được cấu thành từ số lượng
phần tử kém đa dạng. Những vùng này có thể (được) cho điểm cao khiến chương trình nhầm lẫn
1 Thông thường, chuỗi truy vấn nhỏ hơn nhiều so với cơ sở dữ liệu, ví dụ: truy vấn có thể là một nghìn
nucleotide trong khi cơ sở dữ liệu là vài tỷ nucleotide 15 lOMoAR cPSD| 45470709
trong việc tìm các chuỗi thật sự quan trọng trong cơ sở dữ liệu, vì vậy chúng nên được lọc bỏ
ra. Các vùng sẽ được đánh dấu bằng X (trình tự protein) hoặc N (trình tự axit nucleic) và sau đó
bị chương trình BLAST bỏ qua. Để lọc ra các vùng có độ phức tạp thấp, chương trình SEG được
sử dụng cho trình tự protein và chương trình DUST được sử dụng cho trình tự ADN. Mặt khác,
chương trình XNU được sử dụng để che giấu sự lặp lại song song trong chuỗi protein [7].
• Lập danh sách từ gồm k chữ cái của chuỗi truy vấn.
Lấy k = 3 làm ví dụ, nghiên cứu liệt kê các từ có độ dài 3 trong chuỗi protein truy vấn (k
thường là 11 cho một chuỗi ADN) "tuần tự" cho đến khi chữ cái cuối cùng của chuỗi truy vấn.
Phương pháp được minh họa trong hình 2.8.
Hình 2.8. Phương pháp thiết lập danh sách từ truy vấn k-chữ cái [14]
• Liệt kê các cặp chuỗi con tương đồng
Bước này là một trong những điểm khác biệt chính giữa BLAST và FASTA. FASTA quan
tâm đến tất cả các chuỗi con thông thường trong cơ sở dữ liệu và chuỗi truy vấn được liệt kê
trong bước 2.1.1.1; tuy nhiên, BLAST chỉ quan tâm đến những chuỗi con “đạt điểm cao”. Điểm
số được tạo ra bằng cách so sánh chuỗi con trong danh sách ở bước 2.1.1.1 với tất cả các chuỗi
con có 3 chữ cái. Bằng cách sử dụng ma trận cho điểm (ma trận thay thế - substitution matrix)
để cho điểm so sánh của từng cặp, có thể có 203 điểm phù hợp cho một chuỗi con có 3 chữ cái.
Ví dụ, điểm số thu được khi so sánh PQG với PEG và PQA lần lượt là 15 và 12 với sơ đồ trọng
số BLOSUM62. Đối với các chuỗi ADN, một điểm trùng khớp được tính là +5 và điểm không
khớp là -4 hoặc là +2 và -3. Sau đó, ngưỡng điểm (threshold) T được sử dụng để giảm số lượng
các cặp phù hợp có thể có. Các cặp có điểm lớn hơn ngưỡng T sẽ vẫn nằm trong danh sách, trong
khi những cặp có điểm thấp hơn T sẽ bị loại bỏ. Ví dụ, PEG được giữ lại, nhưng PQA bị loại bỏ khi T = 13 [14].
• Sắp xếp các từ đạt điểm cao còn lại thành một cây tìm kiếm hiệu quả. 16 lOMoAR cPSD| 45470709
Điều này cho phép chương trình nhanh chóng so sánh các chuỗi con đạt điểm cao với các
chuỗi con trong cơ sở dữ liệu.
• Lặp lại bước 3 đến bước 4 cho chuỗi con k-chữ cái trong chuỗi truy vấn
• Quét các chuỗi cơ sở dữ liệu để tìm các kết quả khớp chính xác với các từ có điểm cao còn lại
Chương trình BLAST quét các chuỗi trong cơ sở dữ liệu để tìm các chuỗi con có điểm cao
còn sót lại, chẳng hạn như PEG, ở mỗi vị trí. Nếu tìm thấy một cặp chính xác, cặp này được sử
dụng để làm cơ sở cho một liên kết khác giữa các chuỗi truy vấn và chuỗi trong cơ sở dữ liệu.
Mở rộng các cặp tương đồng thành cặp phân đoạn có điểm số cao (High-scoring Segment Pairs - HSP).
• Liệt kê tất cả các HSP trong cơ sở dữ liệu có điểm đủ cao để được xem xét.
• Đánh giá ý nghĩa của điểm HSP.
• Ghép hai hoặc nhiều vùng HSP thành một liên kết dài hơn.
• Hiển thị các sắp xếp Smith-Waterman cục bộ của chuỗi truy vấn và các chuỗi
tương đồng trong cơ sở dữ liệu.
• Báo cáo về các ghép cặp có điểm nhỏ hơn một tham số ngưỡng .
2.2.1.3. Tính điểm của các bắt cặp trình tự và các matrix thay thế
Bắt cặp trình tự BLAST bao gồm một cặp trình tự, trong đó mỗi chữ cái trong một trình
tự được ghép nối với chính xác một chữ cái hoặc một khoảng trống trong chữ cái kia. Điểm bắt
cặp được tính bằng cách gán một giá trị cho từng cặp chữ cái được bắt cặp và sau đó tổng các
giá trị này theo chiều dài của bắt cặp. Đối với sự sắp xếp trình tự protein, điểm cho mọi cặp chữ
cái axit amin có thể được tính ra trong “ma trận thay thế” trong đó các thay thế có khả năng có
giá trị dương và các thay thế không chắc có giá trị âm. Theo mặc định, BLAST sử dụng ma trận
“blosum62”, một thành viên của chuỗi ma trận thay thế được sử dụng phổ biến nhất, tuy nhiên,
một số thành viên của chuỗi PAM cũng có sẵn. Đối với sự liên kết nucleotide,
BLAST tính điểm với +2 cho các cặp chữ cái giống nhau được bắt cặp và −3 cho mỗi cặp chữ
cái được bắt cặp không khác nhau. Việc tạo ra một khoảng trống trong sự liên kết dẫn đến một 17 lOMoAR cPSD| 45470709
điểm trừ "tạo ra khoảng cách", với mỗi phần mở rộng của một khoảng cách đã tồn tại trước đó
sẽ phải bị trừ ít hơn [13].
2.2.1.4. Ý nghĩa thống kê
Các bắt cặp được BLAST tìm thấy được tính điểm, được gán một giá trị thống kê, được
gọi là “Giá trị kỳ vọng”. “Giá trị kỳ vọng” là số lần liên kết tốt hoặc tốt hơn so với giá trị mà
BLAST tìm thấy ngẫu nhiên, dựa trên kích thước của cơ sở dữ liệu được tìm kiếm. Ngưỡng
"Giá trị kỳ vọng" do người dùng đặt, xác định những bắt cặp nào sẽ được báo cáo. Ngưỡng “Giá
trị kỳ vọng” càng lớn thì ít nghiêm ngặt hơn và mặc định BLAST là “10” được thiết kế để đảm
bảo rằng không có liên kết quan trọng về mặt sinh học nào bị bỏ lỡ. Tuy nhiên, “Giá trị kỳ vọng”
trong phạm vi từ 0,001 đến 0,0000001 thường được sử dụng để cho giá trị tốt nhất” [11, 13]. 2.2.2. BioEdit
BioEdit- một chương trình phân tích trình tự sinh học thân thiện với người dùng được cung
cấp miễn phí cho hệ điều hành Windows, cho phép phân tích và chỉnh sửa trình tự.
BioEdit chỉnh sửa trình tự acid nucleotic/protein với đầy đủ các tính năng. BioEdit hỗ trợ đọc
và xử lý nhiều định dạng mở rộng sử dụng trong các ứng dụng tin sinh học khác. Điều này cho
phép hoán đổi các tệp dữ liệu giữa BioEdit và các chương trình khác. Những định dạng đó bao
gồm (định dạng văn bản đa dạng thức * .rtf, fastafiles * .fas * .fasta * .fst * .fsa, tệp ngân hàng
gene * .gbk * .gene * .gb* .gnk, * .csv, phân cách bằng tab, * .txt, excel, abi định dạng tệp sắc
ký đồ, * .ab1 * .abi, tệp trình tự, * .seq, plasmidfiles * .pmd, tệp dự án bioedit * .bio, tệp clustal
* .aln, gcgfiles * .gcg, tệp XML * .xml, tệp acqus, phylib tệp * .phy, tệp NBRF / PIR * .pir, *
.nbf. Có thể dễ dàng bắt đầu một tài liệu mới và sao chép (Ctrl + C) dán (Ctrl + V) dữ liệu [15].
Phần mềm BioEdit được công bố lần đầu tiên vào năm 1999 bởi Tom Hall (Hall, 1999).
Chương trình đã được sử dụng rộng rãi và được trích dẫn trong nhiều bài báo trong tạp chí xếp
hạng. Nhiều nhà khoa học về sinh học phân tử đã sử dụng BioEdit trong các nghiên cứu ban đầu
của họ. Với nhiều trích dẫn trong các tạp chí, BioEdit đã trở thành công cụ tin sinh học quan
trọng của các nhà sinh học phân tử. Ưu điểm: 18 lOMoAR cPSD| 45470709
- Giao diện thân thiện với người dùng và có thể tải xuống trực tuyến miễn phí.
- BioEdit là một chương trình độc lập và có thể áp dụng với nhiều chức năng khác nhau.
- Sử dụng được trên hầu hết các phiên bản Windows. Nhược điểm:
- BioEdit không còn được cập nhật và tài liệu đã lỗi thời.
- Thời gian BioEdit cần để phân tích các trình tự sẽ tăng lên rất nhiều theo độ dài của chúng.
Một số chức năng cần kinh nghiệm xử lý và nhiều bước để thực hiện [15].
2.3 Các nghiên cứu liên quan về ứng dụng dữ liệu geneomic trong tìm kiếm đích tác động
chất kháng nấm ở Candida spp.
2.3.1 So sánh geneomic cho phép xác định các đích tác động của thuốc kháng nấm (Comparative
geneomics allowed the identification of drug targets against human fungal pathogenes) [16]
Đối tượng nghiên cứu:
Nhiên cứu được tiến hành trên Aspergillus fumigatus và Candida albicans, nhóm gene
ban đầu có sẵn trong ngân hàng gene và đang được bảo tồn và cần thiết ở C. albicans và / hoặc
A. fumigatus đã được sử dụngg để xác định 55 ortholog [17] . Ngoài ra, hai gene không thiết
yếu (KRE2 và ERG6) nhưng rất quan trọng đối với khả năng tồn tại của tế bào chủ đã được
thêm vào danh sách các mục tiêu thuốc [18,19]. Sự sắp xếp của 57 trình tự đó so với bộ gene
của 8 loại nấm gây bệnh P. lutzii, P. brasiliensis phân lập được (Pb18 e Pb3), A. fumigatus, B.
dermatitidis, C. albicans, C. Immitis, C. neoformans, H. capsulatum đã xác nhận sự hiện diện
của tất cả các gene. Từ đó chọn ra 10 gene bảo tồn làm mục tiêu thuốc bởi vì chúng có mặt
trong tất cả các loài được phân tích, có trong Candida spp và không có trong bộ gene người (Bảng 2.1). 19
Tài liệu liên quan:
-

Bài giảng sinh học phân tử | Đại học Y Dược Thành phố Hồ Chí Minh
55 28 -

Bài tập sinh di truyền cho môn sinh học phân tử | Đại học Y Dược Thành phố Hồ Chí Minh
61 31 -

Bài 6: Điều Hòa Hoạt Động Gen - Câu Hỏi Sinh Học Phân Tử | Đại học Y Dược Thành phố Hồ Chí Minh
60 30 -

Bài 3 - Các loại ARN: Câu hỏi trắc nghiệm Sinh học phân tử | Đại học Y Dược Thành phố Hồ Chí Minh
61 31 -

Câu hỏi ngắn Sinh học phân tử | Đại học Y Dược Thành phố Hồ Chí Minh
110 55