Bài 2: Số phận của thuốc trong cơ thể | Tài liệu Dược động học
Liệt kê được các quá trình ảnh hưởng đến số phận của thuốc trong cơ thể và biết được ý nghĩa của các thông số dược động học. Liệt kê được các giai đoạn chính trong sự hấp thu thuốc và các yếu tố ảnh hưởng đến sự hấp thu thuốc. Nêu được tính chất của phức hợp thuốc-protein huyết tương trong phân phối thuốc. Tài liệu giúp bạn tham khảo, ôn tập và đạt kết quả cao. Mời đọc đón xem!
Môn: Dược động học(YCT) 5 tài liệu
Trường: Trường Đại học Y dược Cần Thơ 414 tài liệu
Tác giả:














Preview text:
lOMoAR cPSD| 46342985 BÀI 2 DƯỢC ĐỘNG HỌC
SỐ PHẬN CỦA THUỐC TRONG CƠ THỂ
MỤC TIÊU HỌC TẬP
1. Liệt kê được các quá trình ảnh hưởng đến số phận của thuốc trong cơ thể
và biết được ý nghĩa của các thông số dược động học.
2. Liệt kê được các giai đoạn chính trong sự hấp thu thuốc và các yếu tố
ảnh hưởng đến sự hấp thu thuốc.
3. Nêu được tính chất của phức hợp thuốc-protein huyết tương trong phân phối thuốc.
4. Liệt kê được các phản ứng chuyển hóa quan trọng trong chuyển hóa
thuốc và giải thích được cảm ứng và ức chế enzym gan.
5. Kể được và bàn luận về các đường đào thải thuốc quan trọng. NỘI DUNG BÀI HỌC
Dược động học (pharmacokinetics) là phần nghiên cứu những tác động của
cơ thể đến thuốc hay nói cách khác là nghiên cứu về số phận của thuốc trong cơ thể.
Tình trạng của thuốc trong cơ thể là một thuật ngữ rộng, bao gồm các quá
trình hấp thu, phân bố, chuyển hóa và thải trừ - thường được viết tắt như là
ADME (Absorption – Distribution – Metabolism – Elemination). Dược động
học mô tả mối liên hệ giữa liều và nồng độ thuốc tại vị trí tác dụng, và diễn tiến
theo thời gian của nồng độ thuốc trong cơ thể.
1. SỰ HẤP THU THUỐC
Sự hấp thu dược phẩm là quá trình thuốc thâm nhập vào môi trường cơ
thể, đến nơi tác động. Đây là quá trình vận chuyển thuốc từ nơi dùng thuốc
(uống, tiêm,..) vào máu rồi đi khắp cơ thể, đến nơi tác động. Để vào được hệ
tuần hoàn chung của cơ thể, thuốc phải trãi qua 3 giai đoạn hấp thu như sau: ✓
Sự hấp thu ngang qua màng tế bào. ✓
Hiệu ứng vượt qua lần đầu (First-Pass Effect). ✓
Trong hệ tuần hoàn chung. 16 lOMoAR cPSD| 46342985
1.1. Sự hấp thu dược phẩm qua màng tế bào
Dù dùng bất cứ đường cho thuốc nào, để đến các receptor phát sinh hoạt
tính sinh học, thuốc phải vượt vượt qua hàng rào các màng tế bào. Như vậy sự
hấp thu thuốc phụ thuộc rất nhiều vào cấu tạo của màng tế bào. Các chất tan
trong lipid thì dễ dàng di chuyển qua màng. Chính bản chất lipid của màng đã
cản trở sự khuếch tán qua màng của chất tan trong nước, chỉ các chất tan trong
nước có phân tử nhỏ mới khuếch tán được qua màng qua hệ thống các kênh.
1.1.1. Các cơ chế vượt qua màng tế bào của thuốc -
Sự vận chuyển thụ động hay khuyếch tán thụ động, còn gọi là
khuyếch tán đơn thuần hay sự thấm. Thuốc sẽ khuyếch tán từ nơi có nồng độ
cao đến nơi có nồng độ thấp và phụ thuộc vào hệ số phân chia dầu trong nước
(D/N). Hệ số càng cao thì thuốc càng khuyếch tán nhanh qua màng. -
Lọc hay khuyếch tán qua lỗ, di chuyển theo chiều gradien nồng
độ. Quá trình khuyếch tán này được thực hiện bởi các kênh protein cho thấm
qua một cách có chọn lọc một số chất, nhiều kênh có thể mở ra hoặc đóng lại bằng các cổng. -
Sự vận chuyển thuận lợi, là quá trình khuếch tán có sự tham gia
của chất vận chuyển hay chất mang. Sự khuếch tán này di chuyển theo chiều
gradien nồng độ, mang tính đặc hiệu và sẽ đạt trạng thái bão hòa khi chất mang
không còn các vị trí liên kết tự do. -
Sự vận chuyển tích cực hay vận chuyển chủ động, là loại vận
chuyển đặc biệt, thuốc được chuyển qua màng nhờ chất mang nên có thể không
tuân theo định luật Fick và đòi hỏi phải có năng lượng cung cấp. Căn cứ vào
nguồn gốc năng lượng được sử dụng, vận chuyển tích cực được chia thành vận
chuyển tích cực nguyên phát và vận chuyển tích cực thứ phát.
1.1.2. Các yếu tố ảnh hưởng sự hấp thu dược phẩm qua màng tế bào
- Tính chất của dược phẩm
+ Ảnh hưởng của cấu trúc phân tử thuốc: Những thuốc ở dạng phân cực
thường khó khó qua màng, ví dụ các kháng sinh nhóm aminosid. Nhưng các
phân tử thuốc chỉ tan trong lipid lại không qua màng được do không thể tiếp
cận được màng tế bào. 17 lOMoAR cPSD| 46342985
+ Tính hòa tan của dược phẩm: Thuốc ở dạng dung dịch nước dễ hấp thu
hơn dung dịch dầu, dịch treo hay dạng rắn do hòa tan nhanh chóng vào pha nước nơi hấp thu.
+ Điều kiện giúp sự hòa tan thuốc hấp thu dễ dàng:
. Dạng dung dịch, dạng muối Na+, K+
. Kích thước phân tử nhỏ
+ Nồng độ dược phẩm tại nơi hấp thu: Nồng độ càng lớn thì sự hấp thu
càng nhanh đối với các thuốc qua màng bằng khuếch tán qua lớp lipid.
- Đặc điểm nơi hấp thu dược phẩm
+ Tuần hoàn nơi hấp thu: Hệ thống mao mạch nơi hấp thu càng phát triển
thì sự hấp thu thuốc càng dễ dàng.
+ Bề mặt nơi hấp thu: Bề mặt nơi hấp thu càng lớn thì sự hấp thu càng
nhanh như biểu mô phổi, phế nang. Ruột non hấp thu nhanh hơn dạ dày do có
bề mặt hấp thu rộng hơn và lưu lượng máu cao hơn. Dạ dày Ruột non •
Bề mặt hấp thu: 1m2 200 m2 •
Lưu lượng máu: 150ml/ phút 1000ml/ phút
+ Tình trạng nơi hấp thu: Sự hiện diện của thức ăn, tình trạng bệnh lý đường tiêu hóa.
+ Cơ chế làm trống dạ dày. Tốc độ làm rỗng dạ dày chỉ làm thay đổi tốc
độ hấp thu thuốc, chứ không làm thay đổi mức độ hấp thu thuốc. Vì vậy quan
trọng cho những thuốc cần khởi phát tác dụng sớm: thuốc ngủ, thuốc giảm đau. •
Các yếu tố làm tăng co bóp dạ dày có thể làm giảm
hấp thu các thuốc cần môi trường acid dịch vị để hòa tan như tetracyclin, propoxyphen. •
Ngược lại, thuốc bị mất hoạt tính ở dạ dày như PNC,
L.dopa thì kéo dài thời gian làm rỗng dạ dày sẽ làm giãm sinh khả dụng của thuốc.
Để rút ngắn thời gian làm trống dạ dày •
Uống thuốc lúc bụng đói với nhiều nước. •
Giữ bệnh nhân ở vị trí thẳng đứng. •
Dùng thuốc tăng co bóp dạ dày: Metoclopramid, Domperidon. 18 lOMoAR cPSD| 46342985
Để kéo dài thời gian làm trống dạ dày
Uống thuốc lúc bụng no. • Tập thể dục nặng. •
Giữ bệnh nhân ở vị trí nằm. •
Dùng chung với các thuốc giãm co bóp dạ dày: Kháng muscarin, antacid, narcotic.
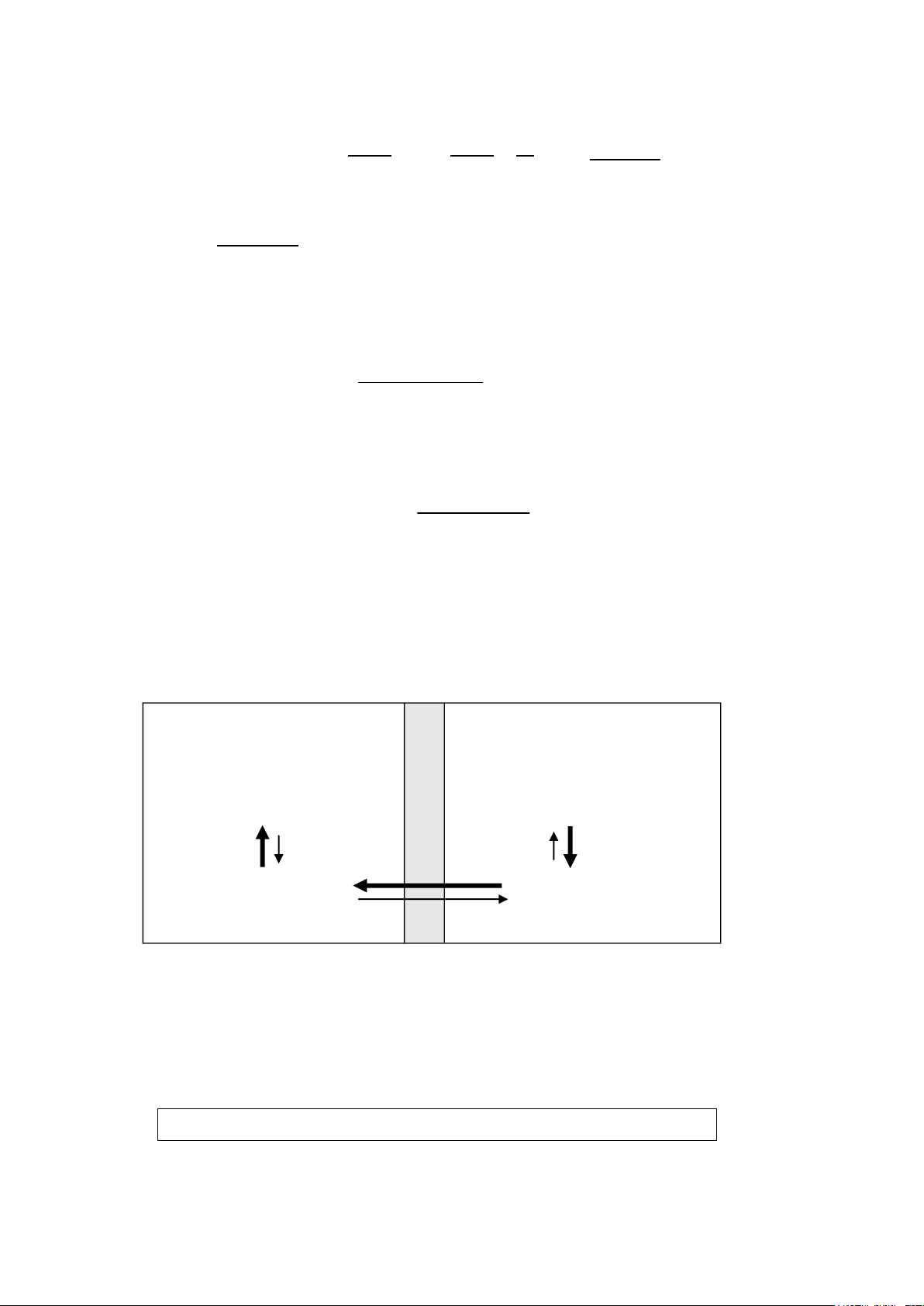
+ pH nơi hấp thu: Đa số các thuốc là acid yếu hoặc base yếu, chúng phân ly theo phương trình sau: ▪ Acid: HA H+ + A- C8H7O2COOH H+ + C8H7O2COO-
Aspirin proton Aspirin anion ▪ Base: B + H2O BH+ + OH- C +
12H11ClN3NH2 + H2O C12H11ClN3NH3 + OH-
Pyrimethamin Pyrimethamin cation
Chỉ có các dạng không ion hóa (HA hoặc B) thì mới dễ dàng khuếch
tán ngang qua màng tế bào (do tính thân trong lipid). Tỷ lệ không ion
hóa/ion hóa của thuốc thì phụ thuộc vào hằng số phân ly của thuốc pKa và
pH của môi trường, thể hiện bằng phương trình Henderson – Hasselbalch.
Theo phương trình Henderson – Hasselbalch, ta có: ▪ Với thuốc là Acid yếu: pH = pKa + log [A -] , suy ra: Ka = [H+] [A-] [HA] [HA] ▪ Với thuốc là Base yếu: + - [BH ] [OH ] pH = pKb + log , suy ra: Kb = [B]
K hằng số phân ly, pKa = - logKa. Ka càng lớn thì pKa càng nhỏ. 19 lOMoAR cPSD| 46342985
▪ Một acid hữu cơ có pKa thấp là một acid hữu cơ mạnh và ngược lại.
▪ Một base có pKa thấp là một base yếu và ngược lại.
Hay nói một cách khác, khi một thuốc có hằng số pKa bằng với pH môi
trường thì 50% thuốc có ở dạng ion hóa và 50% thuốc ở dạng không ion hóa, lúc đó:
Nồng độ phân tử = 1 và log 1 = 0 Nồng độ ion
Bảng 2.1. Trị số pKa và pKb của một số thuốc là acid yếu và base yếu
(ở nhiệt độ 25oC) Acid yếu pKa Base yếu pKb Acid salicylic 3,00 Reserpin 6,6 Acid acetylsalicylic 3,49 Codein 7,9 Sulfadiazin 6,48 Quinin 8,4 Barbital 7,91 Procain 8,8 Acid boric 9,24 Atropin 9,65
Từ phương trình trên, nếu biết pH nơi hấp thu và pKa của thuốc, có thể
tính được tỷ lệ nồng độ:
Nồng độ không ionNồng độ ion hóa hoá
Vì chỉ có dạng không ion hóa mới hấp thu được qua màng nên có thể
biết chất đó dễ hay khó hấp thu và hấp thu theo chiều nào.
❑ Ví dụ về ảnh hưởng của pH trên sự hấp thu thuốc:
Khi uống một thuốc là acid yếu có pKa = 4,4; môi trường dạ dày = 1,4;
môi trường huyết tương = 7,4.
Áp dụng phương trình Henderson – Hasselbalch ta có:
▪ Ở ngăn dạ dày: log [A-] = pH - pKa [AH]
log [RCOO-] = 1,4 – 4,4 = -3 [RCOOH] 20 lOMoAR cPSD| 46342985 [RCOO -] = 1 [RCOOH] 1000 ▪ Ở ngăn huyết tương: log [A-] = pH - pKa [AH]
log [RCOO-] = 7,4 – 4,4 = 3 [RCOOH] [RCOO-] 1000 = [RCOOH] 1
Vì chỉ phần không ion hóa [RCOOH] và có nồng độ cao mới
khuếch tán qua màng cho nên acid này sẽ chuyển từ ngăn dạ dày sang
ngăn huyết tương và được hấp thu. Hàng rào lipid ở ruột
Môi trường huyết tương
Môi trường dạ dày pH = 7 ,4
pH = 1 ,4 pKa = 4 ,4 1000 RCOO - + H +
RCOO - + H + 1 1 RCOOH
RCOOH 1000
❑ Sự ion hóa của thuốc còn phụ thuộc vào pH môi trường
Bảng 2.2. Ảnh hưởng của pH đến sự ion hóa của acid salicylic có pKa = 3
pH Phần trăm (%) không ion hóa 21 lOMoAR cPSD| 46342985 1 99,0 2 90,9 3 50,0 4 9,09 5 0,99 6 0,10
Như vậy acid salicylic được hấp thu nhiều ở dạ dày. Qua bảng trên cho thấy
khi bị ngộ độc thuốc, muốn ngăn sự hấp thu thuốc hay tăng sự đào thải thuốc
đã hấp thu ra ngoài cơ thể, ta thay đổi pH của môi trường.
❑ Ảnh hưởng của pH dịch vị
Tại dạ dày, pH acid là điều kiện cần thiết để một số thuốc có thể hấp thu được
khi xuống ruột. Vì vậy việc sử dụng đồng thời các chất giảm tiết HCl dịch vị
có thể làm giảm hấp thu các thuốc dùng chung. Mặt khác pH acid dạ dày cũng
làm tăng khả năng hòa tan một số thuốc có bản chất kiềm, nhờ đó tạo thuận lợi
hơn cho khả năng thuốc hấp thu tại ruột.
Tuy nhiên, độ acid cao ở dạ dày cũng phá hủy đi một số thuốc kém bền trong
môi trường acid (như ampicillin, erythromycin,…); một số thuốc sẽ chuyển
sang dạng ion hóa nên gây bất lợi cho hấp thu, ví dụ như các PPI (omeprazol,
pantoprazol) do rã trong dạ dày nên nhận thêm 1 proton H+ trở nên mang điện
tích, không thể hấp thu qua được niêm mạc ruột.
Bảng 2.3. Một số kháng sinh hấp thu rất kém khi đưa qua đường uống Kháng sinh Tên nhóm thuốc Kháng sinh nhóm aminosid
Amikacin, gentamicin, neomycin, streptomycin
Kháng sinh nhóm Beta-lactam Carbenicillin, cefazolin, cefotaxim, ceftazidim Kháng sinh khác Vancomycin, Teicoplanin
Cần lưu ý một số thuốc thực chất có hấp thu tại dạ dày, nhưng do thuốc chỉ lưu
lại ở dạ dày một thời gian ngắn và bề mặt dạ dày thì không thuận lợi cho sự hấp
thu bằng bề mặt niêm mạc ruột non, nên tỷ lệ hấp thu thì không đáng kể khi so
với tại ruột non. Ví dụ ở bảng 2.2, theo lý thuyết thì lẽ ra dạ dày (pH=3) sẽ là
nơi hấp thu thuận lợi cho acid salicylic (là sản phẩm chuyển hóa của aspirin)
khi sử dụng đường uống. Nhưng thực tế ruột non lại là vị trí hấp thu thuận lợi
hơn do có độ tưới máu nhiều, bề mặt niêm mạc rộng nên làm cho khả năng hấp 22 lOMoAR cPSD| 46342985
thu thuốc tại ruột non nhanh hơn dạ dày nhiều. Do đó, việc kiểm soát tốc độ
làm rỗng dạ dày cũng là một khâu quan trọng trong sử dụng thuốc theo đường uống.
- Các yếu tố khác: + T h ứ c ă n + T u ổ i t á c + Bệnh lý + Tương tác thuốc
+ Dạng thuốc, thành phần, công thức của chế phẩm.
1.2. Hiệu ứng vượt qua lần đầu (First-pass effect)
- Hiệu ứng vượt qua lần đầu là sự mất đi một lượng thuốc do các enzyme
của một cơ quan chuyển hóa thuốc ngay lần đầu tiên khi thuốc tiếp xúc với
cơ quan này. Thành phần thuốc bị biến đổi gọi là chất chuyển hóa.
- Để đánh giá hiệu ứng vượt qua lần đầu, người ta sử dụng hệ số ly trích
(ER: The extraction ratio).
ER được định nghĩa là tỷ lệ lượng thuốc hấp thu bị ly trích
(nghĩa là bị bắt giữ lại ở cơ quan hoặc bị mất đi) ở cơ quan
chuyển hóa do hiệu ứng vượt qua lần đầu trước khi thuốc vào
đến hệ tuần hoàn. Hệ số ER thay đổi từ 0 (không bị ly trích)
đến 1 (có sự ly trích hoàn toàn) tùy theo loại thuốc sử dụng.
1.2.1. Hiệu ứng vượt qua lần đầu ở ruột. 23 lOMoAR cPSD| 46342985 - Thuốc màng nhầy ruột Tĩnh mạch cửa gan.
- Enzym ở màng nhầy ruột thực hiện các phản ứng chuyển hóa thuốc
làm mất đi một lượng thuốc trước khi thuốc vào hệ tuần hoàn chung.
- Chức năng và hậu quả:
+ Tạo chất dễ tan trong nước và dễ đào thải qua nước tiểu
+ Mất đi một phần thuốc sử dụng làm giảm hoạt tính trị liệu của thuốc
- Hệ số ly trích ở ruột (ERI): là tỉ lệ lượng thuốc hấp thu bị ly trích ở
ruột do hiệu ứng vượt qua lần đầu trước khi thuốc vào đến hệ tuần hoàn.
- Một số thuốc chịu hiệu ứng vượt qua lần đầu ở ruột:
Alpha-methyl dopa Dexamethason Flurazepam Isoprenalin Pethidin Ppropoxyphen
Pentazocin Sulfamid Metoclopramid Chlopromazin
1.2.2. Hiệu ứng vượt qua lần đầu ở gan.
- Thuốc gan tĩnh mạch trên gan tim, phổi.
- Gan chứa hầu hết các enzyme tham gia chuyển hóa thuốc với số lượng
và hoạt tính cao, do đó sự thất thoát thuốc do chuyển hóa thường xảy ra chủ yếu ở gan.
- Chức năng và hậu quả:
+ Chức năng: tạo chất dễ tan trong nước và dễ đào thải qua nước tiểu
+ Hậu quả: mất đi một lượng thuốc trước khi vào hệ tuần hoàn
chung, làm giảm họat tính trị liệu thuốc.
- Hệ số ly trích ở gan (ERH): là tỉ lệ lượng thuốc hấp thu bị ly trích ở
gan do hiệu ứng vượt qua lần đầu của phần thuốc được hấp thu và vượt qua
màng nhầy ruột trước khi thuốc vào đến hệ tuần hoàn.
- Một số thuốc chịu hiệu ứng vượt qua lần đầu ở gan:
Aspirin Lidocain Petazocin Morphin Cortison
Hexobarbital Isoprenalin Propoxyphen Propranolol Metoprolol
1.2.3. Hiệu ứng vượt qua lần đầu ở
phổi - Thuốc phổi hệ tuần hoàn chung.
- Phản ứng enzyme phổi: oxydase.
- Chức năng và hậu quả: phổi là nơi cuối cùng mà một thuốc có thể bị
thất thóat trước khi vào hệ tuần hoàn chung. 24 lOMoAR cPSD| 46342985
- Hệ số ly trích ở phổi (ERP): là tỉ lệ bị ly trích ở phổi do hiệu ứng vượt
qua lần đầu của phần thuốc đã được hấp thu và vượt qua sự chuyển hóa ở ruột và gan.
- Một số thuốc chịu hiệu ứng vượt qua lần đầu ở phổi, sau khi được IV: Chlorpromazin Imipramin Methadon Methadon Isoprenalin Nortriptylin
1.3. Trong hệ tuần hoàn chung – Một số thông số dược động học liên
quan đến quá trình hấp thu
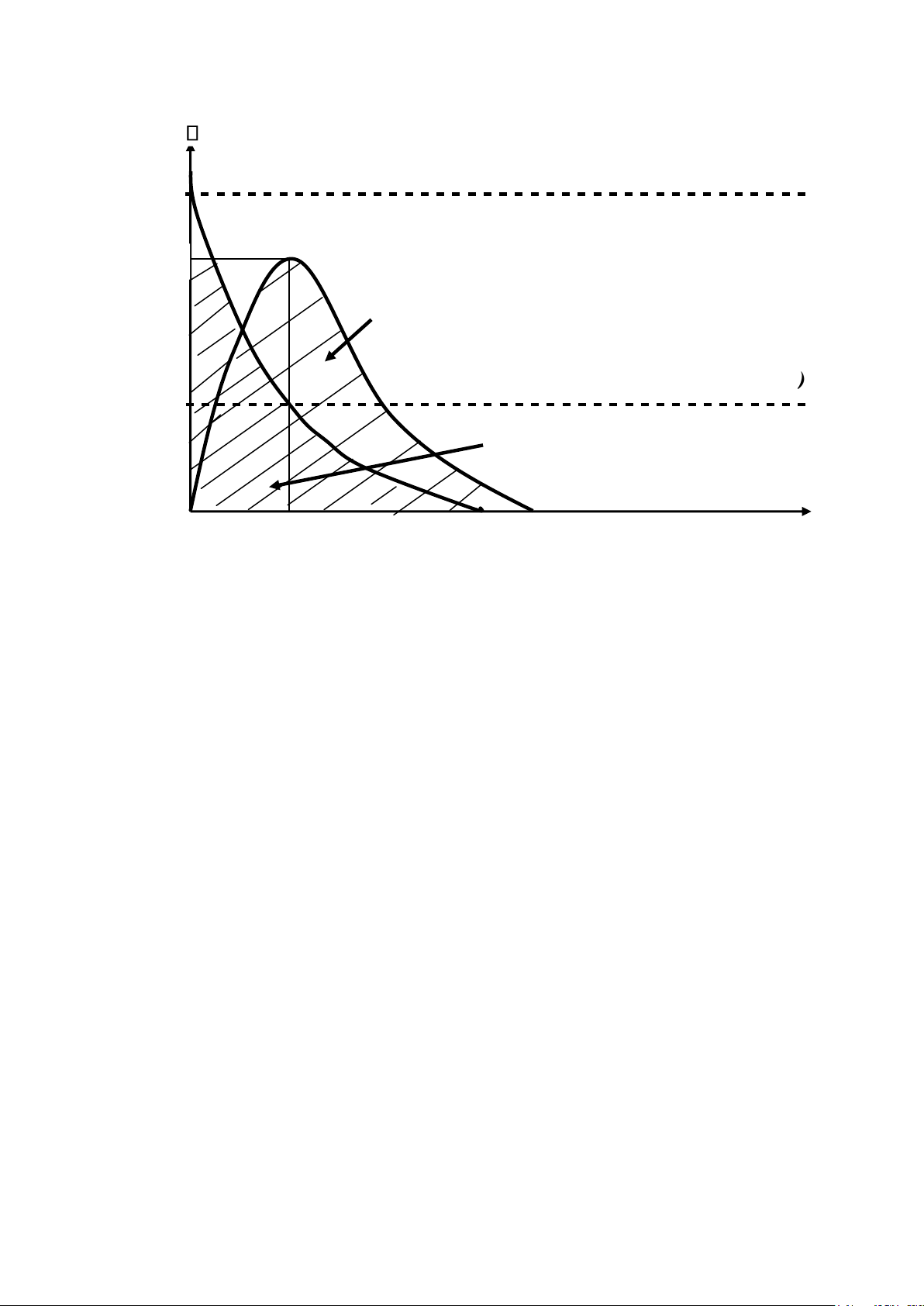
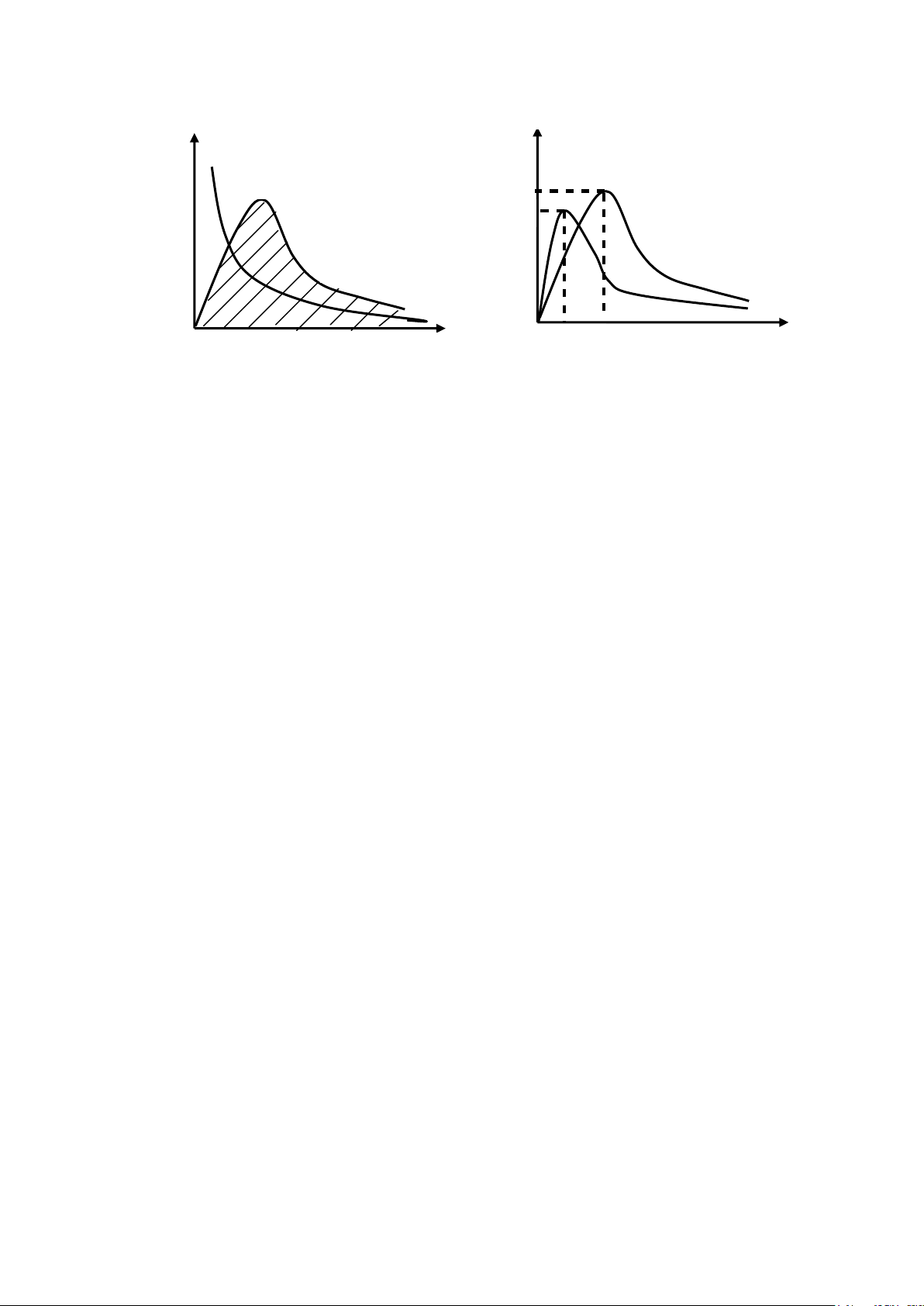
1.3.1. Diện tích dưới đường cong biểu diễn nồng độ – thời gian
(AUC: Area Under the Curve). ✓
Cmax: nồng độ thuốc tối đa đạt được trong huyết tương (đánh giá cường độ tác động) ✓
Tmax: thời điểm thuốc đạt được nồng độ tối đa (đánh giá tốc độ
hấp thu) ✓ Đơn vị tính AUC là mg.h.l -1 hoặc g.h.ml-1.
Sau khi đưa thuốc vào cơ thể (tiêm, uống), máu được lấy ở các
thời điểm khác nhau để xác định nồng độ thuốc trong huyết tương
(Cp), sẽ có được diện tích dưới đường cong AUC (đồ thị biểu diễn sự
biến thiên của nồng độ thuốc trong máu theo thời gian). AUC biểu
thị tượng trưng cho lượng thuốc vào được vòng tuần hoàn ở dạng còn
hoạt tính sau những khoảng thời gian t nhất định.
Để tính AUC, người ta có thể sử dụng nguyên tắc hình thang, hoặc phương pháp
tích phân. Từ giá trị của AUC nồng độ-thời gian, có thể tính được trị số sinh
khả dụng của thuốc, AUC phản ánh mức độ hấp thu dược chất. 25 lOMoAR cPSD| 46342985 C (
g/l )
Nồng đ ộ tối thiểu gây độc (MTC) C
ma x
AUC của thuốc đưa theo đường uống
Nồng độ tối thiểu có hiệu lực ( ) MEC
AUC của thuốc đưa theo đường IV Tmax t ( h)
Hình 2.1. Đồ thị nồng độ thuốc trong huyết tương theo thời gian sau
khi dùng một liều duy nhất
1.3.2. Khái niệm về sinh khả dụng (SKD; Bioavailability, F%)
Thực tế khi sử dụng thuốc cho bệnh nhân, không phải tất cả lượng thuốc
được dùng đều phát huy tác dụng, mà chỉ một phần nào đó có tác dụng, vào
được vòng tuần hoàn ở dạng nguyên vẹn, chưa bị chuyển hóa. Phần thuốc này
được gọi là sinh khả dụng của thuốc, ký hiệu là F (Fraction of dose), đơn vị tính là %.
▪ Định nghỉa Sinh khả dụng: Sinh khả dụng của thuốc là thông
số biểu thị tỷ lệ (%) lượng dược chất vào vòng tuần hoàn chung ở dạng còn
hoạt tính (chưa bị chuyển hóa) so với liều đã dùng (D0), tốc độ (Tmax) và
cường độ (Cmax) thuốc thâm nhập được vào vòng tuần hoàn chung.
Đây là một trong số các thông số chính của dược động học và đặc trưng
cho pha hấp thu của thuốc. Sinh khả dụng được đặc trưng bởi phần khả dụng F và vận tốc hấp thu.
1.3.2.1. Phần khả dụng F (Mức độ khả dụng)
Trị số F dùng để ước tính số lượng thuốc có trong cơ thể. Nếu thuốc được
đưa vào cơ thể bằng đường tĩnh mạch thì F=1. Các thuốc được đưa vào bằng
đường ngoài tĩnh mạch luôn có F<1 do bởi:
+ Thuốc được hấp thu không hoàn toàn.
+ Bị chuyển hóa ở đường tiêu hóa, máu, gan. 26 lOMoAR cPSD| 46342985
+ Tái hấp thu không hoàn toàn qua chu trình gan-ruột.
+ Biến đổi sinh học ở gan.
Trị số F chỉ được đánh giá trong mối tương quan với một dạng bào chế
quy chiếu. Có thể so sánh SKD tương đối của hai chế phẩm cùng loại hoạt chất,
cùng hoặc khác hảng bào chế cho trên cùng một nhóm bệnh nhân.
1.3.2.2. Vận tốc hấp thu (Tốc độ khả dụng) Được
đánh giá bởi 3 yếu tố:
+ Nồng độ tối đa trong huyết tương (Cmax).
+ Thời gian để đạt được nồng độ tối đa (Tmax). +
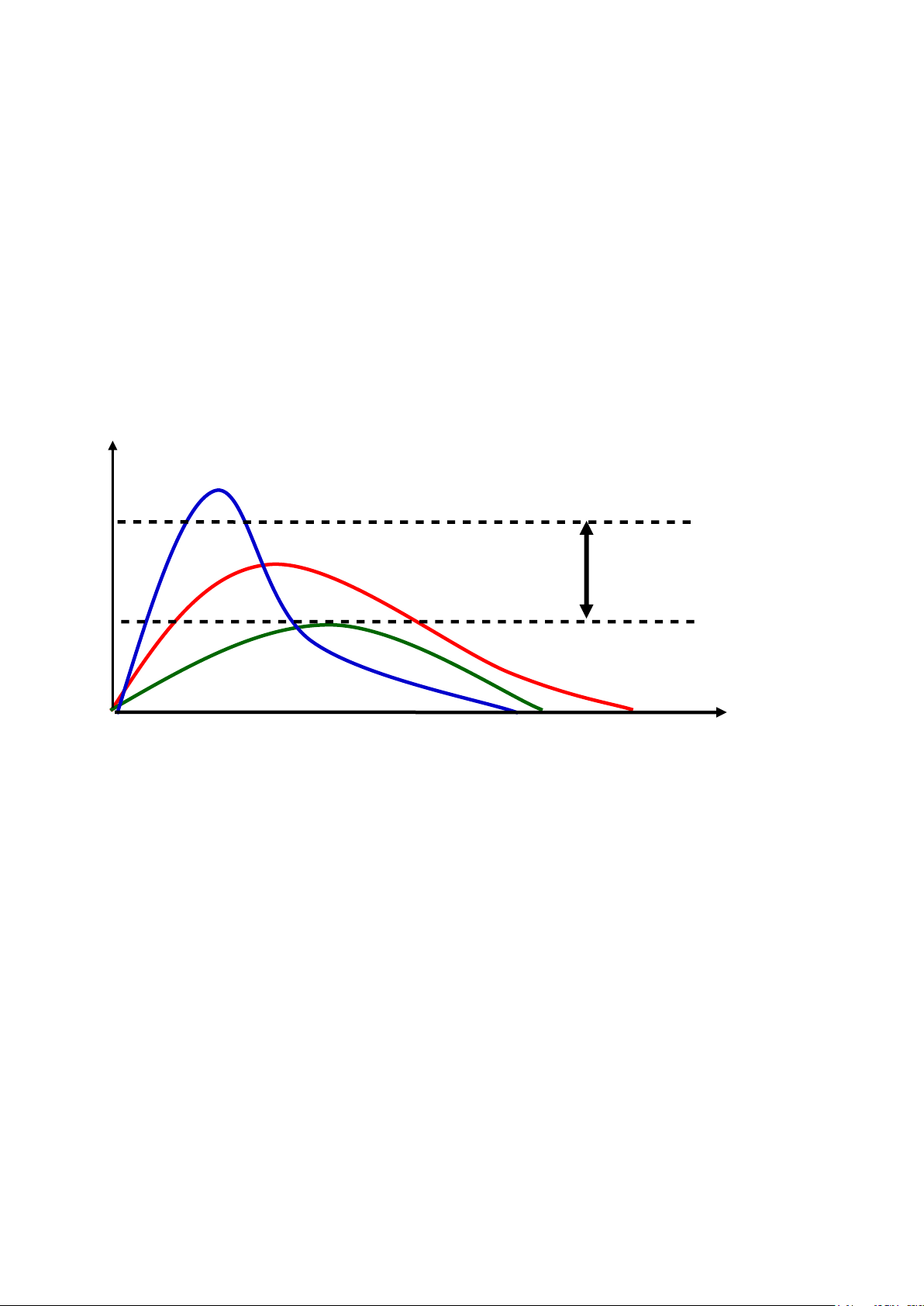
Hằng số của vận tốc hấp thu (Ka). C (
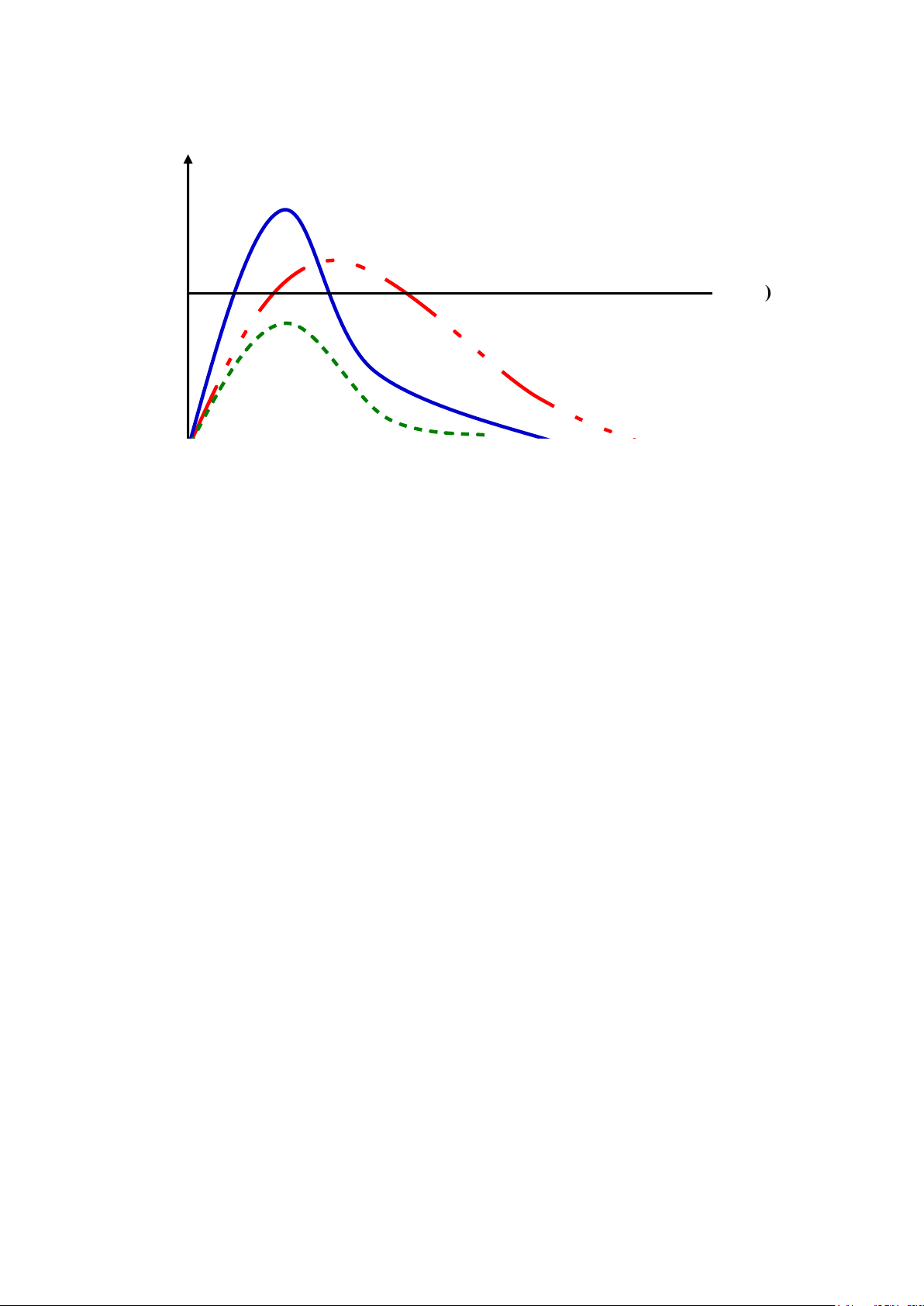
µ g/l) MTC
Thuố c A Khoảng điều trị Thuốc B ME C Thuốc C t(h)
Hình 2.2. Đồ thị biễu diễn nồng độ thuốc trong huyết tương theo thời gian lấy mẫu
máu của 3 dạng bào chế A, B và C của cùng một hoạt chất
Theo đồ thị biễu diễn trên, AUC thuốc A = AUC thuốc B = AUC thuốc C , cho
thấy lượng thuốc vào máu là như nhau. Tuy nhiên, do tốc độ hấp thu hoạt chất
khác nhau nên hiệu quả sẽ điều trị khác nhau.
Sự khác biệt về tốc độ khả dụng sẽ quan trọng đối với thuốc dùng một lần (như thuốc gây mê). 27 lOMoAR cPSD| 46342985
C (µg/l) Thuốc A (M E ) C Thuốc B Thuốc C t (h ) Hình
2.3 . Đồ thị biễu diễn nồng độ thuốc trong huyết tương theo thời gian để
so sánh SKD của 3 chế phẩm A, B và C
Theo đồ thị biễu diễn trên:
▪ Có sự khác biệt về hiệu lực lâm sàng với 3 chế phẩm A, B và C: A và C
được hấp thu vào máu với tốc độ bằng nhau (cùng Tmax), trong khi đó AUC
của A và B thì tương đương nhau và lớn hơn AUC của C.
▪ Liều A đạt nồng độ hiệu lực tối thiểu sớm hơn liều C và duy trì với thời
gian dài hơn (nên quan trọng đối với thuốc chỉ dùng một lần).
▪ Tuy vậy, khi dùng chế độ nhiều liều, cả liều A và C sẽ phát sinh cùng
một nồng độ trung bình trong máu.
1.3.2.3. Tương đương sinh học và sinh khả dụng
Theo định nghĩa, để có được SKD thì phải tính được lượng thuốc vào được
vòng tuần hoàn. Tuy nhiên không thể thực hiện được, do các quá trình dược
động (quá trình ADME) của thuốc xảy ra cùng một lúc, nghĩa là lượng thuốc
vào vòng tuần hoàn sẽ biến động liên tục theo thời gian. Vì vậy cần phải sử
dụng thông số AUC để hổ trợ tính toán.
Có 2 loại sinh khả dụng:
+ Sinh khả dụng tuyệt đối (F% tuyệt đối, F1): là tỉ lệ giữa trị số AUC
thu được khi đưa thuốc ngoài đường tĩnh mạch (thông thường là đường uống)
so với trị số AUC đưa qua đường tĩnh mạch của cùng một dược chất.
Ý nghĩa: Dùng để đánh giá ảnh hưởng của đường sử dụng trên hiệu quả
sinh học của dược chất. Nếu so sánh giữa 2 liều bằng nhau: 28 lOMoAR cPSD| 46342985
F% tuyệt đối = (AUCt ) PO x 100% (AUCt ) IV
Nếu dùng khác liều, công thức điều chỉnh sẽ là:
F = (AUCt ) PO x D IV x 100% (AUCt ) IV x DPO
Giá trị SKD cho sẵn hoặc trong tính toán thường là giá trị của SKD tuyệt đối.
+ Sinh khả dụng tương đối (F’ hay F2): là tỷ lệ so sánh giữa 2 giá trị
SKD của cùng một hoạt chất, cùng một đường đưa thuốc, cùng một mức liều
nhưng của hai hãng sản xuất thuốc khác nhau hoặc của hai dạng bào chế khác
nhau. Thông thường SKD tương đối là tỷ lệ giữa hai giá trị SKD của hai dạng
bào chế khác nhau của cùng một thuốc dùng qua đường uống.
F’ = (AUCt ) TEST(hãng A) x 100% (AUCt ) STANDARD (hãng B) Hay
F’ = ( AUC(AUCt ) tT )HUỐC ĐỐI CHIẾU THUỐC THỬ x 100%
Nếu dùng khác liều, ta có công thức điều chỉnh
(AUCt ) test x Dstandard
F’ = x 100%(AUCt ) standard x Dtest
Nếu chế phẩm thử có SKD = 80-125% so với chế phẩm đối chiếu thì
được coi là tương đương sinh học với chế phẩm đối chiếu. 29 lOMoAR cPSD| 46342985 Nồng độ Nồng độ IV C max C max PO PO T max T max Thời gian T hờ i gian
Sinh khả dụng tuyệt đối
Sinh khả dụng tương đối
Hình 2.4. Sinh khả dụng tuyệt đối và sinh khả dụng tương đối +
Tương đương sinh học (TĐSH, Bioequivalence, BE):
TĐSH là khi không có sự khác nhau có ý nghĩa về tốc độ và mức độ sẵn
có tại nơi tác dụng của cùng một dược chất hoặc chất có tác dụng từ hai thuốc
sau khi được dùng cùng một mức liều ở cùng một điều kiện thử nghiệm trong
một nghiên cứu được thiết kế phù hợp.
Đánh giá SKD tương đối thực chất là xác định tương đương sinh học
(TĐSH). Tiêu chí đánh giá TĐSH là những thông số cơ bản của dược động học
thể hiện mức độ và tốc độ hấp thu của thuốc: F%, Tmax và Cmax. Thông
thường tỷ lệ cho phép dao động trong khoảng 20%. Với những thuốc có phạm
vi điều trị hẹp, TĐSH chỉ đạt được nếu cả 3 thông số: AUC, Cmax và Tmax
bằng nhau (sai khác nhau trong phạm vi cho phép).
Thực hiện TĐSH là nhằm so sánh thuốc generic với thuốc của hảng phát
minh hoặc thuốc có uy tín trên thị trường, nhằm giúp người làm công tác điều
trị lựa chọn được đúng chế phẩm thay thế. Nếu hai chế phẩm tương đương bào
chế thì chưa đủ thay thế trong trị liệu; chỉ có những chế phẩm TĐSH với nhau
mới được dùng thay thế cho nhau trong điều trị.
Vì vậy hai chế phẩm là TĐSH khi:
+ Là hai chế phẩm tương đương bào chế hay thay thế dược học có SKD
không khác nhau có ý nghĩa thống kê, hay nói cách khác là hai chế phẩm có
AUC, Cmax và Tmax không khác nhau.
+ Mức độ hấp thu (AUC, Cmax) không khác nhau, sự khác nhau về tốc
độ hấp thu (Tmax) do cố ý, được ghi trong nhãn, không quan trọng với việc đạt
nồng độ trị liệu trong trường hợp điều trị bệnh mạn tính và được xem không có ý nghĩa lâm sàng. 30 lOMoAR cPSD| 46342985
+ Múc độ khác biệt được chấp nhận là không quá 20%, hoặc có thể thay
đổi tuỳ phương pháp đánh giá hay xử lý thống kê. 1.3.2.4. Ý nghĩa của SKD và TĐSH.
+ Là cơ sở để lựa chọn chế phẩm: SKD tuyệt đối của thuốc uống đạt
>50% là có thể chấp nhận được, nếu >80% thì coi như sự hấp thu thuốc vào
máu qua đường uống xấp xỉ đường IV; nếu <50% thì dạng uống khó đạt yêu
cầu trị liệu trong bệnh nặng. Ví dụ Ampicillin và Amoxicillin đều có cùng phổ
tác dụng, nhưng SKD của ampicillin chỉ 30-50% trong khi amoxicillin là 60-
90% nên được ưu tiên lựa chọn cho đường uống.
+ Là cơ sở để lựa chọn đường cho thuốc: Với thuốc có SKD qua đường
uống trên 80% thì coi như hấp thu tương tự đường IV, nên chỉ tiêm tĩnh mạch
khi không uống được. Ví dụ các kháng sinh nhóm quinolon có SKD >80% nên
đường uống là chọn lựa ưu tiên.
+ Giá trị SKD tương đối cho biết khả năng thay thế trong điều trị: Khi
các thông số đặc trưng là AUC, Cmax và Tmax của thuốc thử và thuốc đối
chứng khác nhau nằm trong phạm vi cho phép (thường từ 80-125% tính theo
giá trị log) thì coi như thuốc thử tương đương với thuốc đối chứng và có thể
thay thế nhau trong điều trị. Khi đăng ký thuốc generic để xin cấp phép lưu
hành, TĐSH phải là việc đánh giá bắt buộc.
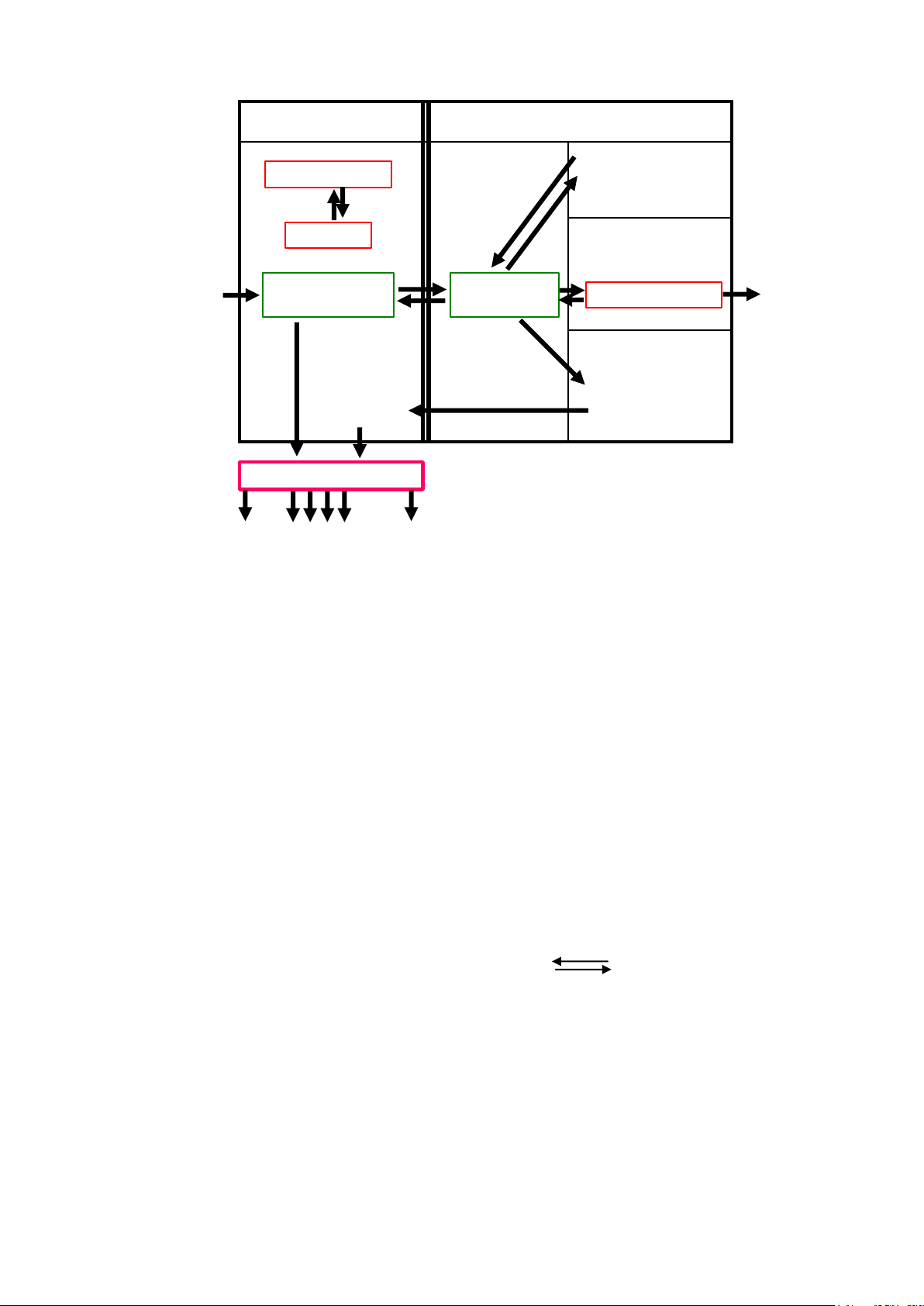
2. SỰ PHÂN PHỐI DƯỢC PHẨM
Sau khi được hấp thu vào tuần hoàn chung, dược phẩm sẽ phân bố khắp cơ
thể. Trong cơ thể, dược phẩm thường hiện diện ở 2 dạng: -
Dạng tự do có khả năng phát sinh ra hiệu ứng dược lý. -
Dạng gắn kết được vận chuyển và phân phối trong cơ thể. Sự phân
phối của thuốc trong cơ thể phụ thuộc vào: - Tính tan trong lipid của thuốc. -
Lưu lượng máu tới mô - Thành phần hóa học của mô. 31 lOMoAR cPSD| 46342985
HUY Ế T TƯƠNG MÔ
Thu ố c - protein
NƠI D Ự TR Ữ Protein
NƠI TÁC D Ụ NG THU
Thu ố c
Thu ố c Ố C Thu (D ( D TÁC
ố c - receptor
ạ ng t ự
ạ ng t ự D do) do)
Ụ NG
NƠI CHUY Ể N HOÁ
Ch ấ t
Ch ấ t chuy ể n
chuy ể n hoá hoá
Ờ NG TH Ả I TR Ừ
Thận Đường khác Tiêu hoá
Hình 2.5. Quá trình vận chuyển thuốc trong cơ thể (theo E.Singlas)
2.1. Sự phân bố trong máu – sự gắn kết với protein huyết tương Sau
khi vào tuần hoàn chung, thuốc có thể ở dưới 2 dạng: -
Dạng tự do tan trong huyết tương có khả năng phát sinh ra hiệu ứng dược lý. -
Dạng gắn kết với các thành phần của máu là protein và hồng cầu
được vận chuyển và phân phối trong cơ thể.
2.1.1. Sự hình thành phức hợp thuốc-protein huyết tương
Trong máu, thuốc sẽ gắn kết với các protein có trong huyết tương, hình thành
một phức hợp dược phẩm-protein huyết tương. K1
[Thuốc] + [Protein huyết tương] [Thuốc-Protein huyết tương] Dạng tự do
K2 Dạng phức hợp
Với K1 và K2 là hằng số tốc độ phối hợp và phân ly của phức hợp
thuốcprotein huyết tương. 2.1.2. Tính chất
Dạng phức hợp dược phẩm-protein huyết tương có những tính chất sau: 32 lOMoAR cPSD| 46342985
- Không có tính chuyên biệt, nhiều thuốc khác nhau có thể gắn trên cùng
một vị trí của protein huyết tương.
- Khi còn ở dạng phức hợp thì dược phẩm không sinh tác động dược lực,
không bị chuyển hóa và đào thải.
- Có tính thuận nghịch trong gắn kết, phức hợp được xem là một tổng kho
dự trữ thuốc trong cơ thể.
- Giữ một chức năng đệm hiệu quả, đảm bảo cho sự cân bằng giữa lượng
dược phẩm bị gắn kết với lượng dược phẩm ở dạng tự do đủ gây tác dụng dược lực.
- Có sự cạnh tranh giữa những dược phẩm có cùng ái lực với một loại protein.
- Khả năng hình thành phức chất này rất kém ở trẻ sơ sinh.
Sự gắn kết vào protein huyết tương của thuốc được biểu thị bằng tỷ lệ gắn
kết f hay fu, với:
- f là tỷ lệ nồng độ thuốc gắn vào protein huyết tương so với nồng độ thuốc toàn phần.
f = [Thuốc gắn vào protein huyết tương] [Thuốc toàn phần]
- fu là tỷ lệ nồng độ thuốc tự do trong huyết tương so với nồng độ thuốc toàn phần.
Các loại protein tham gia gắn kết với thuốc là:fu = 1 – f
- Albumin: chiếm 50-60% protein huyết tương, có vai trò chính yếu trong
phản ứng gắn thuốc và có nhiều điểm gắn trên phân tử. Thường gắn chủ
yếu các thuốc là acid yếu. - Globulin.
- -1 - glycoprotein acid: thường gắn các thuốc là kiềm yếu.
- Lipoprotein: gắn với một lượng nhỏ thuốc kiềm.
Tỷ lệ gắn kết thay đổi tùy theo dược phẩm. Dựa trên tỷ lệ này, người ta có thể phân loại:
- Các thuốc gắn kết mạnh (> 75%)
- Các thuốc gắn kết trung bình (35% - < 75%)
- Các thuốc gắn kết yếu (< 35%)
Các yếu tố ảnh hưởng đến sự gắn kết thuốc với protein là: 33 lOMoAR cPSD| 46342985
- Số vị trí trên protein (n).
- Nồng độ của các protein gắn kết với thuốc (P).
- Hằng số ái lực Ka, biểu thị lực gắn kết.
Bảng 2.4. Tỷ lệ gắn kết với protein huyết tương của một số thuốc
Thuốc là acid yếu Thuốc là base yếu 75-100% Phenylbutazon Diazepam Warfarin Digitoxin Phenytoin Chlopromazin Aspirin Erythromycin 35-<75% Benzylpenicillin Chloroquin Methotrexat Morphin < 35% Ethosuximid Isoniazid
2.1.3. Ý nghĩ của sự gắn kết vào protein huyết tương -
Trong điều trị, đối với thuốc gắn mạnh vào protein huyết tương,
cần dùng liều tấn công cao để bảo hòa các vị trí gắn và đến liều duy trì mới đạt hiệu quả mong muốn. -
Ở trẻ sơ sinh, nhất là thiếu tháng, khả năng gắn kết vào protein rất
kém nên trẻ sơ sinh nhạy cảm với nhiều thuốc. -
Khi dự trữ protein huyết tương giảm (bệnh cấp tính, có thai, chấn
thương, phỏng, suy kiệt), dạng tự do tăng dần lên và độc tính tăng theo, nên cần phải giảm liều. -
Do có hiện tượng cạnh tranh gắn kết, nên chất có ái lực mạnh sẽ
đẩy chất có ái lực yếu ra khỏi vị trí gắn kết và có thể gây độc tính do tăng nồng
độ thuốc tự do trong máu. Ví dụ, phenylbutazol đẩy tolbutamid gây hạ đường
huyết quá mức. Tuy vậy, hiện tượng cạnh tranh gắn kết trên protein chỉ có ý
nghĩa lâm sàng khi thuốc bị đẩy ra khỏi protein huyết tương có những tính chất sau:
+ Thể tích phân phối Vd thấp. 34 lOMoAR cPSD| 46342985
+ Có hệ số trị liệu thấp hay chỉ số trị liệu hẹp (thuốc kháng đông
đường uống, hạ đường huyết uống, chống động kinh, trị ung thư, trị loạn nhịp
tim, kháng sinh nhóm aminosid).
+ Khởi đầu tác động nhanh phụ thuộc nồng độ thuốc trong máu.
+ Thuộc loại gắn mạnh vào protein huyết tương nên ở dạng tự do với
tỷ lệ nhỏ. Ví dụ 97% Dicoumarol gắn vào protein huyết tương, khi bị
phenylbutazol đẩy khỏi huyết tương khoảng 3% thì tác động chống đông tăng lên 100%.
2.2. Sự phân bố và tích lũy ở mô 2.2.1. Định nghĩa
Sự phân bố tại mô là quá trình phân phối thuốc vào trong toàn bộ các mô
và cơ quan. Số lượng thuốc phân phối trong cơ thể tùy vào sinh khả dụng của
thuốc. Từ huyết tương, dược phẩm sẽ được phân phối vào nhiều mô khác nhau. Khi vào trong các mô:
- Thuốc có thể gắn vào các thụ thể chuyên biệt (receptor) để cho tác động dược lực.
- Thuốc cũng có thể gắn vào các điểm nhận (aceptor) để được dự trữ ở mô.
- Hoặc thuốc được gắn vào các enzym để bị chuyển hóa.
Như vậy, tác động dược lực chỉ thể hiện ở những mô có chứa các thụ thể chuyên biệt đối với thuốc.
Một số dược phẩm có ái lực với các mô cao hơn với protein của huyết tương
nên sau khi phân phối vào các mô, thuốc tích lũy hẳn trong các mô này và ít
được đào thải ra khỏi cơ thể. Ví dụ :
- Tetracyclin gắn kết chặt trên hệ xương.
- Aminoglycosid tập trung ở mô thận và tai trong. Nguyên nhân của sự tích luỷ:
- Do có sự tương hợp chặt chẽ về cấu trúc giữa dược phẩm và mô. - Do sự vận chuyển chủ động.
2.2.2. Các yếu tố ảnh hưởng đến nồng độ thuốc ở mô - Sự
tưới máu ở các cơ quan hay lưu lượng máu đến mô.
- Đặc tính lý hóa của thuốc. - Khuynh độ nồng độ.
- Ái lực của thuốc đối với protein của mô và protein của huyết tương.
- Mức độ gắn kết vào protein huyết tương. 35