DNA Topoisomerases - Tài liệu môn Sinh học phân tử | Đại học Huế
DNA Topoisomerases - Tài liệu môn Sinh học phân tử | Đại học Huế. Tài liệu được sưu tầm giúp bạn tham khảo, ôn tập và đạt kết quả cao. Mời bạn đọc đón xem.
Môn: Sinh học phân tử ( DHS) 9 tài liệu
Trường: Đại học Huế 411 tài liệu
Tác giả:




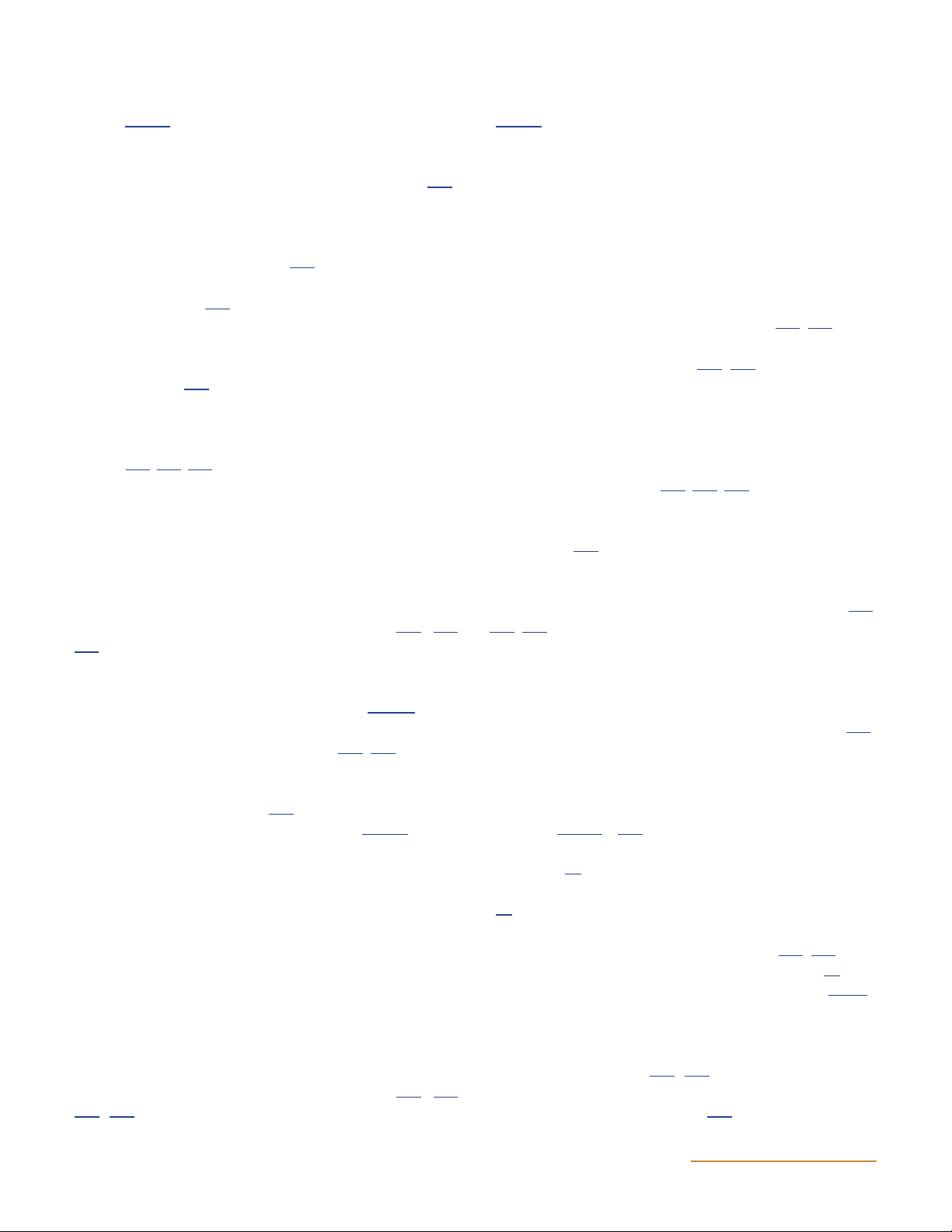

Preview text:
DOMAIN 4 SYNTHESIS AND PROCESSING OF MACROMOLECULES DNA Topoisomerases
NATASSJA G. BUSH,1 KATHERINE EVANS-ROBERTS,2 AND ANTHONY MAXWELL,1
1Department of Biological Chemistry, John Innes Centre, Norwich NR4 7UH, United Kingdom
2Babraham Institute Enterprise Ltd., Babraham Research Campus, Cambridge CB22 3AT, United Kingdom
ABSTRACT DNA topoisomerases are enzymes that control the topology of DNA in
all cells. There are two types, I and II, classified according to whether they make
transient single- or double-stranded breaks in DNA. Their reactions generally involve
the passage of a single- or double-strand segment of DNA through this transient break,
stabilized by DNA-protein covalent bonds. All topoisomerases can relax DNA, but DNA
gyrase, present in all bacteria, can also introduce supercoils into DNA. Because of
their essentiality in all cells and the fact that their reactions proceed via DNA breaks,
topoisomerases have become important drug targets; the bacterial enzymes are key
targets for antibacterial agents. This article discusses the structure and mechanism
of topoisomerases and their roles in the bacterial cell. Targeting of the bacterial
topoisomerases by inhibitors, including antibiotics in clinical use, is also discussed. Received: 1 October 2014 Accepted: 9 October 2014
“Since the two chains in our model are intertwined, it is essential for them to untwist if Posted: 17 April 2015
they are to separate…. Although it is difficult at the moment to see how these processes
Supercedes previous version: http://www
occur without everything getting tangled, we do not feel that this objection will be
.asmscience.org/content/journal/ecosalplus insuperable.” /10.1128/ecosalplus.4.4.9
J. D. WATSON AND F. H. C. CRICK
Editor: Susan T. Lovett, Brandeis University, Waltham, MA
Citation: EcoSal Plus 2015; doi:10.1128/
This quotation, taken from the second Watson and Crick paper detailing the ecosalplus.ESP-0010-2014.
structure of DNA (1), predicts a potential problem inherent in the double-
Correspondence: Anthony Maxwell: tony.
helical structure. The processes that utilize DNA, such as transcription, rep- maxwell@jic.ac.uk
lication, and recombination, require either the temporary or permanent
Copyright: © 2015 American Society for
Microbiology. All rights reserved.
separation of the complementary strands of the double helix. The structure
doi:10.1128/ecosalplus.ESP-0010-2014
of duplex DNA inevitably leads to topological consequences, such as the
introduction of supercoils, during these processes. These changes in topology
are resolved by members of a ubiquitous family of enzymes known as DNA
topoisomerases (2, 3, 4, 5, 6, 7). Topoisomerases alter DNA topology by
binding to the DNA, cleaving either one or both strands of the double
helix, then (for most of these enzymes) passing either the other strand of
the same helix or another double strand through the break, and finally
resealing the DNA backbone. DNA cleavage always involves the formation
of a transient phosphodiester bond between one end of the broken strand
and a tyrosine in the active site of the topoisomerase. Some topoisomerases
require divalent metal ions as cofactors in the DNA cleavage-religation ASMScience.org/EcoSalPlus 1 Bush et al.
reaction. The reactions performed by DNA topoisomer-
specific role is manipulation of DNA topology. Similarly,
ases are depicted in Fig. 1 and Fig. 2. It should be pointed
there may be enzymes currently classified as topoisom-
out that many enzymes (e.g., ligases and recombinases)
erases (e.g., topoisomerase III [topo III] and reverse
can affect DNA topology but are not referred to as topo-
gyrase) whose principal cellular function may be another
isomerases, which is a term reserved for enzymes whose activity.
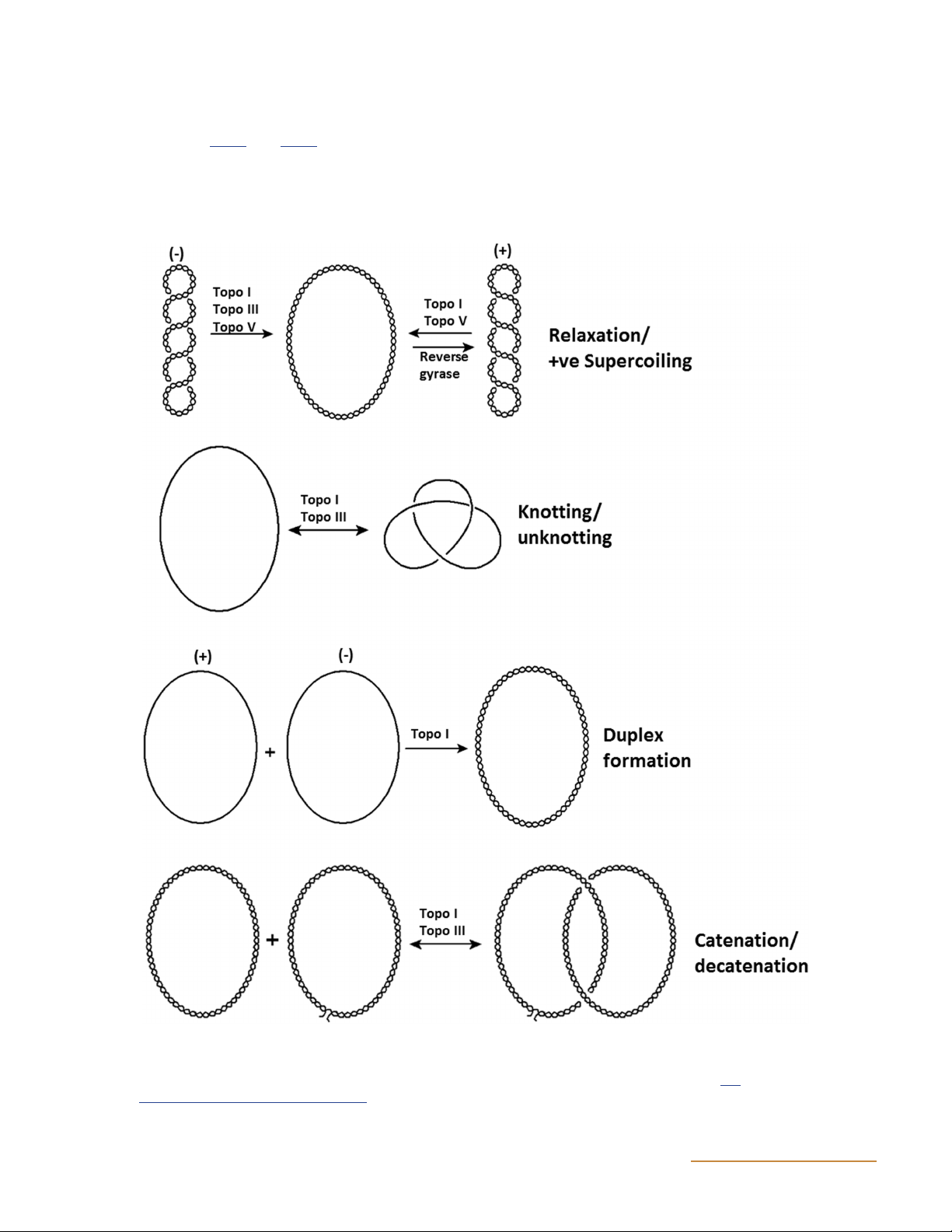
Figure 1 Reactions performed by type I topoisomerases. Examples of specific type I topoisomerases that catalyze the indicated reactions are
given above the arrows. It is important to note that in the decatenation/catenation reaction, the non-nicked plasmid may be supercoiled before
decatenation/catenation occurs; for illustrative purposes it has been drawn as relaxed. (Adapted from reference 313 with permission of the
publisher.) doi:10.1128/ecosalplus.ESP-0010-2014.f1 2 ASMScience.org/EcoSalPlus DNA Topoisomerases
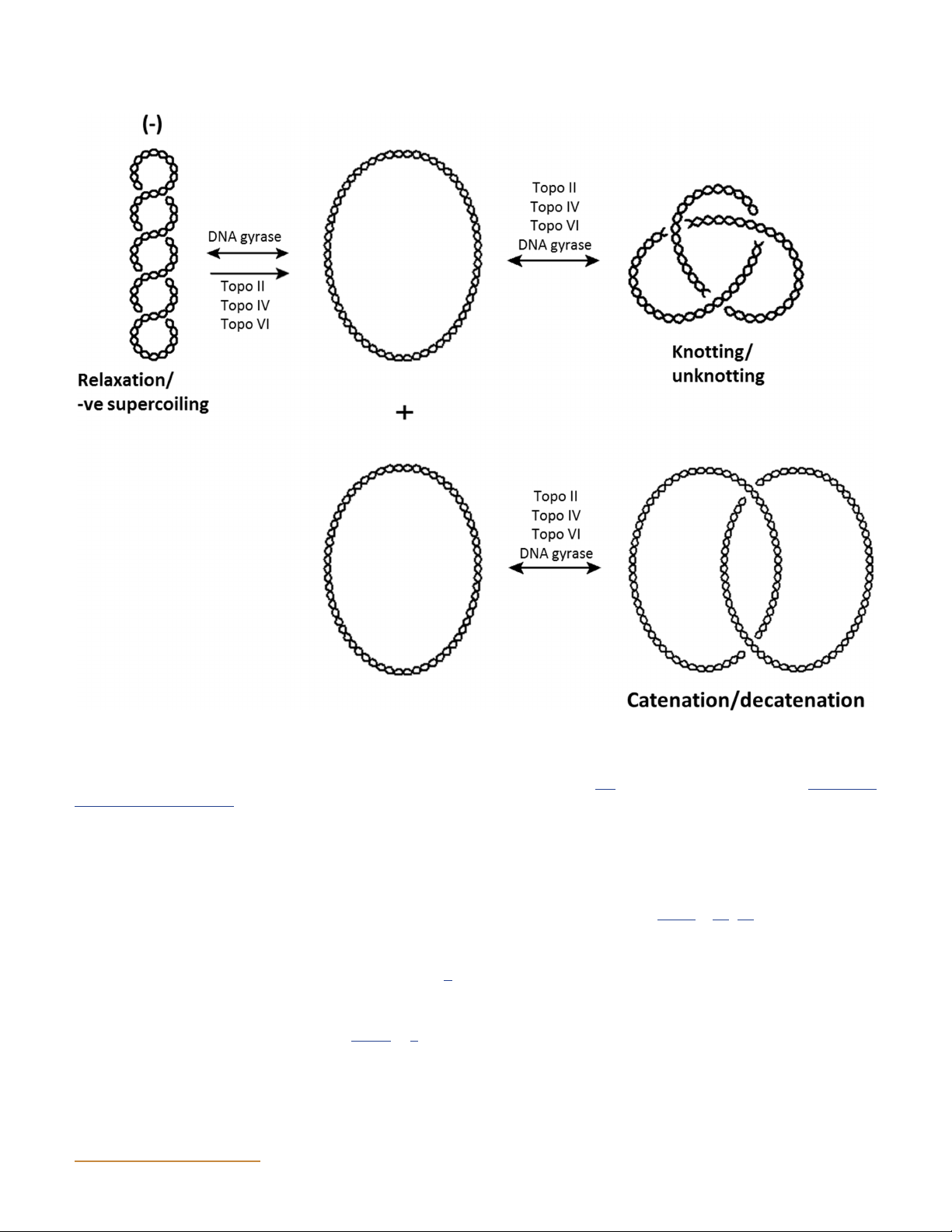
Figure 2 Reactions performed by type II topoisomerases. Examples of specific type II topoisomerases that catalyze the indicated reactions are
given above the arrows. It is important to note that in the decatenation/catenation reaction, the plasmids may be supercoiled before decatenation/
catenation occurs; for illustrative purposes they have been drawn as relaxed. Although only relaxation of negative supercoils is shown, all known
type II topoisomerases can relax positively supercoiled DNA as well. (Redrawn from reference 313 with permission of the publisher.) doi:10.1128/ ecosalplus.ESP-0010-2014.f2
DNA supercoiling can be either positive (correspond-
When two replication forks converge at the end of DNA
ing to over-twisting of the helix) or negative (corre-
replication, catenated DNA rings can be formed if the
sponding to under-twisting of the helix). The binding
daughter molecules are interwound; i.e., precatenanes are
of proteins to DNA is often dependent on the DNA
converted to catenanes (Fig. 4) (10, 11). These rings can
being negatively supercoiled; initiation of the replica-
be separated by decatenation, in which one DNA ring is
tion of bacterial plasmids requires negative supercoiling
cleaved and the other ring is passed through the double-
to facilitate the unwinding of the origin sequence (8).
strand break. As discussed below, this situation can be
As DNA replication proceeds, positive supercoils are
resolved by the activities of various topoisomerases.
generated ahead of the replication fork and so-called
precatenanes may build up behind it (Fig. 3) (9). The
Transcription may also result in changes to DNA topol-
supercoils are removed by topoisomerases to prevent ex-
ogy, for example, if the DNA is anchored to a fixed point
cess supercoiling and the breakdown of the replication
in the cell. It has been proposed that during transcrip- machinery.
tion DNA rotates on its axis to allow RNA polymerase ASMScience.org/EcoSalPlus 3 Bush et al.
DNA single strands, whereas type II enzymes introduce
transient double-strand breaks (19). The two types of
topoisomerases can be further subdivided into type IA,
IB, IC, IIA, and IIB enzymes according to structural,
mechanistic, and evolutionary considerations. The prop-
erties of the different groups of topoisomerases are listed
in Table 1, which summarizes the in vitro activities of
the different enzymes. Alignments of the domains of
the type I and type II topoisomerases are shown in Fig. 5 and Fig. 6, respectively.
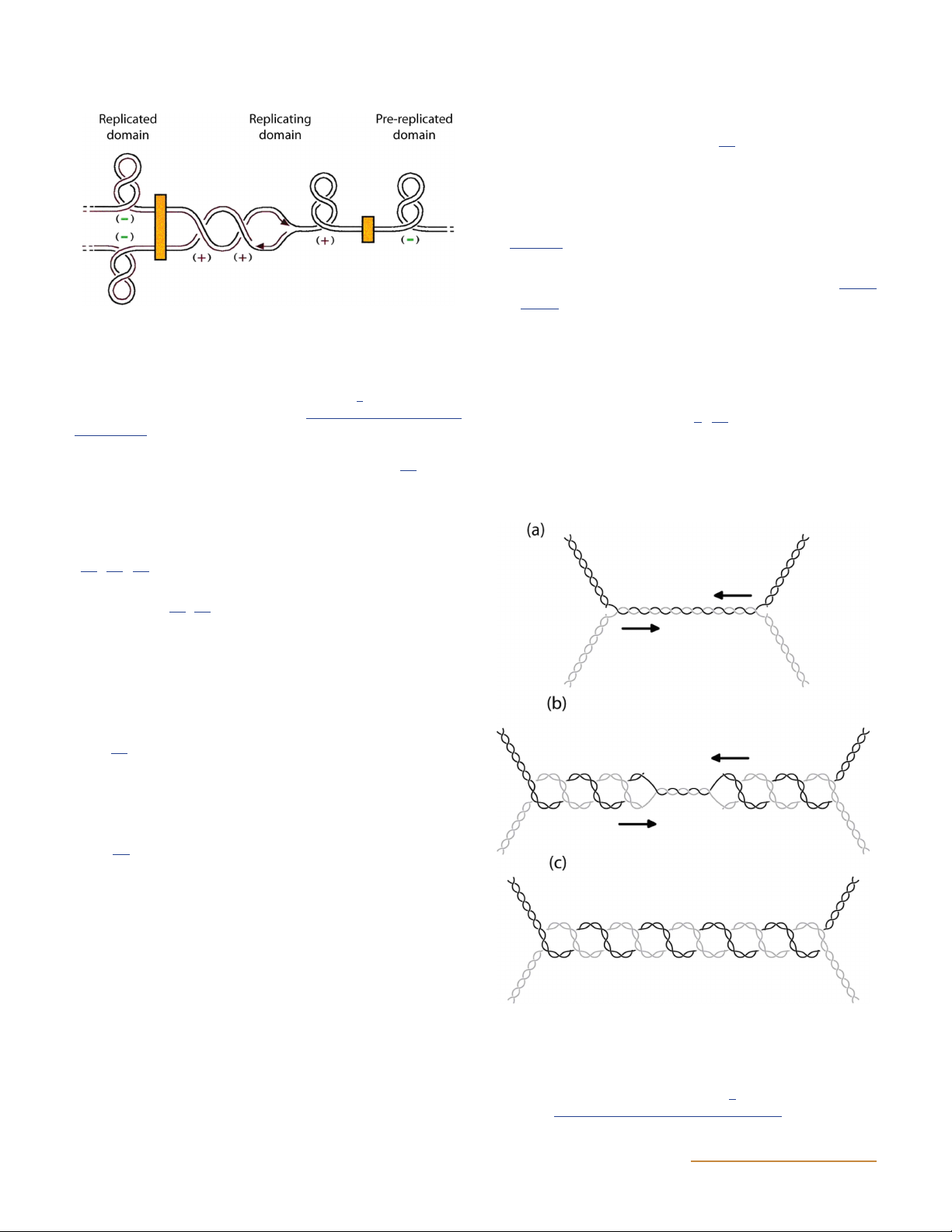
Figure 3 Model of the topology of a replicating chromosome. The
chromosome is separated into domains with the boundaries rep- Type I Topoisomerases
resented as orange boxes; the replication fork is in the center. Positive Topo I
supercoiling occurs ahead of the replication fork, and precatenanes
may form behind it. (Reprinted from reference 9. Copyright 2001
Topo I was the first topoisomerase discovered and was
National Academy of Sciences, U.S.A.) doi:10.1128/ecosalplus.ESP-
originally named ω protein (6, 20). It is found in both 0010-2014.f3
prokaryotes and eukaryotes and can relax negative
to follow the helical path of the DNA strands (12). This
supercoils and catenate and decatenate nicked DNA
rotation leads to the buildup of positive supercoils ahead
of the transcription complex and negative supercoils
behind it, and in prokaryotes these supercoils can be
removed by the enzymes DNA gyrase, topo IV and topo I
(11, 13, 14). The transcription of many genes has been
shown to be influenced by the level of supercoiling in the
bacterial cell (15, 16). This is because supercoiling affects
DNA binding by RNA polymerase and other proteins
that repress or activate transcription. In fact, the levels of
transcription of the genes encoding topo I (topA) and
DNA gyrase (gyrA and gyrB) are all affected by the degree
of supercoiling, in what is thought to be a homeostatic
mechanism to control the amount of supercoiling in the
cell (17). Increased negative supercoiling increases the
transcription of topA and decreases the expression of
gyrA and gyrB. Although its major role is thought to be in
decatenation, topo IV has also been shown to participate
in supercoiling control by contributing to DNA relaxa- tion (18).
In this article we discuss the structures, roles, and mecha-
nisms of the different topoisomerases. We also describe
compounds that inhibit topoisomerases. Although this
review focuses mainly on prokaryotic topoisomerases,
some details of eukaryotic topoisomerases are also pro- vided for comparison.
Figure 4 Formation of catenated DNA at the termination of replica-
tion. (a and b) Converging replication forks (a) lead to the inter-
CLASSIFICATION OF TOPOISOMERASES
winding of daughter molecules and the formation of precatenanes (b).
(c) Upon the completion of replication, the products are catenated
DNA topoisomerases can be divided into two classes:
DNA circles. (Reprinted from reference 2 with permission of the
type I topoisomerases introduce transient breaks into
publisher.) doi:10.1128/ecosalplus.ESP-0010-2014.f4 4 ASMScience.org/EcoSalPlus DNA Topoisomerases
(21). Bacterial topo I enzymes (e.g., Escherichia coli
topo I) are type IA enzymes (Table 1) and can relax +ve No No No No No No No No Yes
only negatively supercoiled DNA. Eukaryotic topo I
enzymes are type IB enzymes (Table 1) and can relax Supercoiling −ve No No No No No No No Yes No
both positively and negatively supercoiled DNA; they b
are evolutionarily and mechanistically distinct from +ve No Yes Yes No Yes Yes Yes Yes No
the bacterial enzymes. E. coli topo I is a 97-kDa pro- b
tein consisting of three domains (22). The first (N- Relaxation −ve Yes Yes Yes Yes Yes Yes Yes Yes Yes
terminal) domain, which consists of 582 amino acids
in E. coli, is responsible for cleavage and strand passage Activities
and contains the active-site tyrosine at position 319. c c
The next 162 amino acids make up a Zn(II)-binding Knotting/ unknotting Yes Yes Yes Yes Yes Unknown Unknown Yes No
domain that contains three tetracysteine motifs. The
C-terminal third domain, which contains 121 amino
acids, is rich in basic amino acids and contributes to substrate binding. c c c d Catenation/ decatenation Yes Yes Yes Yes Yes Unknown Yes Yes No
An N-terminal 67-kDa fragment of E. coli topo I was the
first type I topoisomerase structure to be solved (22). The
structure forms a “base” and a “lid” around a cavity with Mg(II) dependent Yes No Yes Yes Yes No Yes Yes Yes
a diameter of 28 Å, which could accommodate double-
stranded DNA. The active-site tyrosine is positioned at
the entrance to this cavity. Since then, other structures
of type I topoisomerases have been solved, including ATP dependent No No Yes No Yes No Yes Yes Yes
the structure of the catalytic domain of E. coli topo I in
a covalent complex with bound DNA (Fig. 7) (23). Such
information is very valuable in terms of the design of new passage passage passage passage passage passage passage (IC). s antibiotics (see below). clas A. Proposed mechanism Strand Controlled rotation Strand Strand Strand Controlled rotation Strand Strand Strand DN In the proposed new
“enzyme-bridging” model of DNA re- ′3 a
laxation by topo I, the enzyme cleaves a single strand ′or ′ ′ ′ ′ ′ ′ ′ ′ ′ 5 bond formed 5 3 5 5 5 3 5 5 5 form
of DNA and bridges the gap through which the intact to supercoiled
strand is passed (22). The clamp then closes around DNA
the intact strand, and the cleaved strand is religated. The of sitively
protein then reopens to release the passed strand and proposed po No. strands cleaved 1 1 2 1 2 1 2 2 1 ed.
closes again to complete the cycle (Fig. 8). Recent work been ates
suggests that the intact strand may bind in the clamp has strand t indic le
prior to cleavage and then pass through the enzyme- bu topoisomerases +ve sing
stabilized single-strand break (24). IB A; or weak. type DN is Enzyme structure Monomer Monomer Homo-dimer Monomer Hetero-tetramer Monomer Hetero-tetramer Hetero-tetramer Monomer
Mutations in Salmonella enterica serovar Typhimurium as nicked different ed gyrase a is
(Salmonella Typhimurium) and E. coli topo I enzymes of rcoiled A
(topA mutations) are generally nonlethal but lead to Type IA IB IIA IA IIA IB/IC IIB IIA IA describ supe DN li
the acquisition of compensatory mutations in the DNA substrate ively co E.
gyrase genes, gyrA and gyrB (25, 26, 27, 28); however, iginally one properties negat if by
topA mutations have been shown to lead to cold sen- or s
sitivity (28). Despite the apparently nonessential na- Key Bacterial Eukaryotic was only gyrase V le
ture of topo I, the stabilization of the complex of topo I 1 I II III IV V VI gyrase indicate
and cleaved DNA induces the SOS response, lead- Topo -ve Possib Decatenation Topoisomerase Topo Topo Topo Topo Topo Topo DNA Reverse a b c d
ing to cell death (29). There is, therefore, potential Table ASMScience.org/EcoSalPlus 5 Bush et al.
Figure 5 Primary domain structures of type I topoisomerases. Black bars indicate catalytic residues. Y is the catalytic tyrosine which forms the
covalent bond with the phosphodiester backbone of the cleaved single-strand of DNA (319 in E. coli topo I, 328 in E. coli topo III, 809 in
A. fulgidus reverse gyrase, 723 in human topo I, and 226 in M. kandleri topo V) (for a full description of all catalytic residues, see reference 147).
In type IB, NTD is the N-terminal domain, CTD is the C-terminal domain. In type IC, HTH is helix-turn-helix, HhH is helix-hairpin-helix.
(Adapted from reference 148. Schoeffler AJ, Berger JM. 2008. DNA topoisomerases: harnessing and constraining energy to govern chromosome
topology. Q Rev Biophys 41:41–101. © Cambridge University Press, reproduced with permission.) doi:10.1128/ecosalplus.ESP-0010-2014.f5
to develop antibacterial compounds targeted to topo I Topo III
(30, 31); indeed, compounds leading to the stabiliza-
Topo III is a type IA topoisomerase that relaxes and
tion of the topo I cleavage complex have been isolated
decatenates DNA but also has the ability to cleave and (32).
decatenate RNA molecules (37, 38). Topo III is well con-
served across evolutionary lineages and is found in pro-
In contrast to prokaryotic topo I, eukaryotic topo I is
karyotes, eukaryotes, and archaea, but this discussion is
capable of relaxing both positively and negatively super-
limited to the prokaryotic enzyme. Topo III has signifi-
coiled DNA (33). In the proposed mechanism for eu-
cant homology to E. coli topo I, and its crystal structure
karyotic topo I (originally proposed for vaccinia virus
is also very similar (Fig. 7), with four distinct domains
topo I), a single strand of the DNA is broken following
(37, 39). Topo III deletion mutants are viable, so it is
DNA binding (34). The 5′-OH of the broken strand
thought that the enzyme shares in vivo activities with
can then rotate around the other strand before the break
other topoisomerases (37). A topo III (topB) deletion mu-
is resealed, in a process known as controlled rotation
tation in a topo IV temperature-sensitive background is
(35). Human topo I has become a well-established target
lethal (40), which may point to the ability of topo III
for anticancer chemotherapy, with camptothecin and
to decatenate. One difference between the structures of
analogs being widely used clinically (36).
E. coli topo III and topo I is the presence of an additional 6 ASMScience.org/EcoSalPlus DNA Topoisomerases
Figure 6 Primary domain structures of type II topoisomerases. Black bars indicate catalytic residues. Y is the catalytic tyrosine which forms the
covalent bond with the phosphodiester backbone of the cleaved strand of DNA (782 in S. cerevisiae topo II, 122 in E. coli DNA gyrase, 120 in
E. coli topo IV, and 105 in M. mazei topo VI) (for full description of all catalytic residues, see reference 148). GHKL is the ATPase domain,
TOPRIM stands for topoisomerase/primase domain, WHD is the winged-helix domain, CTD is the C-terminal domain, H2tH is the helix-helix-
turn helix domain, and Ig is an immunoglobulin-type fold (not seen in all species). (Adapted from reference 148. Schoeffler AJ, Berger JM. 2008.
DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q Rev Biophys 41:41–101. © Cambridge University
Press, reproduced with permission.) doi:10.1128/ecosalplus.ESP-0010-2014.f6
loop in topo III known as the decatenation loop. Topo I is
with RecQ helicases by resolving stalled and converging
able to decatenate only singly catenated molecules (21),
replication forks (47), such as in the process illustrated in
whereas topo III can unlink multiple catenated dimers Fig. 4.
(41). This, along with the evidence that the deletion of the
loop greatly reduces the decatenation activity, indicates
that the decatenation loop provides topo III with the Topo V
ability to carry out multiple decatenation reactions (41).
Topo V has been described as a type IB topoisomerase
Although topo III is capable of relaxing DNA, this does
(48) as it shows similarities to eukaryotic topo I and can
not appear to be its primary function in E. coli. It has
relax negatively and positively supercoiled DNA. How-
been shown to be involved in the resolution of precate-
ever, it is now thought to be a member of its own class
nanes and in the segregation of chromosomes during
of topoisomerases, type IC, based on structural and
replication (40, 42). RecQ helicases are often linked to
biochemical analyses (49, 50). The crystal structure of
topo III, and the two types of enzyme may function in
topo V identified a novel fold, and it appears to have a
cooperation to resolve some recombination intermedi-
different positioning of the active-site tyrosine, suggest-
ates (43, 44, 45, 46); however, they can also function in-
ing a different mechanism for the cleavage-religation re-
dependently of each other in E. coli (42). Topo III is also
action (50). To date it has been found in only one genus
proposed to have a role in maintaining genome stability
of Archaea (Methanopyrus); it was initially discovered ASMScience.org/EcoSalPlus 7 Bush et al.
helicase-like domain (58, 59) that contains an ATP-
binding site, and there is evidence that this is the domain
that binds DNA (60). If the helicase domain is deleted,
relaxation of negative supercoils can take place in the
absence of ATP, which is typical of type 1A topoiso-
merases (61). Accordingly, if the topoisomerase domain
is deleted, the remaining helicase domain can unwind
DNA transiently in an ATP-dependent reaction. In ad-
dition, DNA unwinding has been shown with full-length
reverse gyrase (62). It has been proposed that the con-
trolled unwinding of DNA by reverse gyrase ensures that
strand passage occurs in the direction of positive super-
coiling (62, 63). As positively supercoiled DNA is more
likely than negatively supercoiled DNA to be resistant
to the harmful effects of high temperature, it is likely
that the action of reverse gyrase is an adaptation to
the extreme habitat occupied by the hyperthermophilic archaea. Type II Topoisomerases
Type II topoisomerases occur in both prokaryotes and
eukaryotes, and these enzymes show a number of simi-
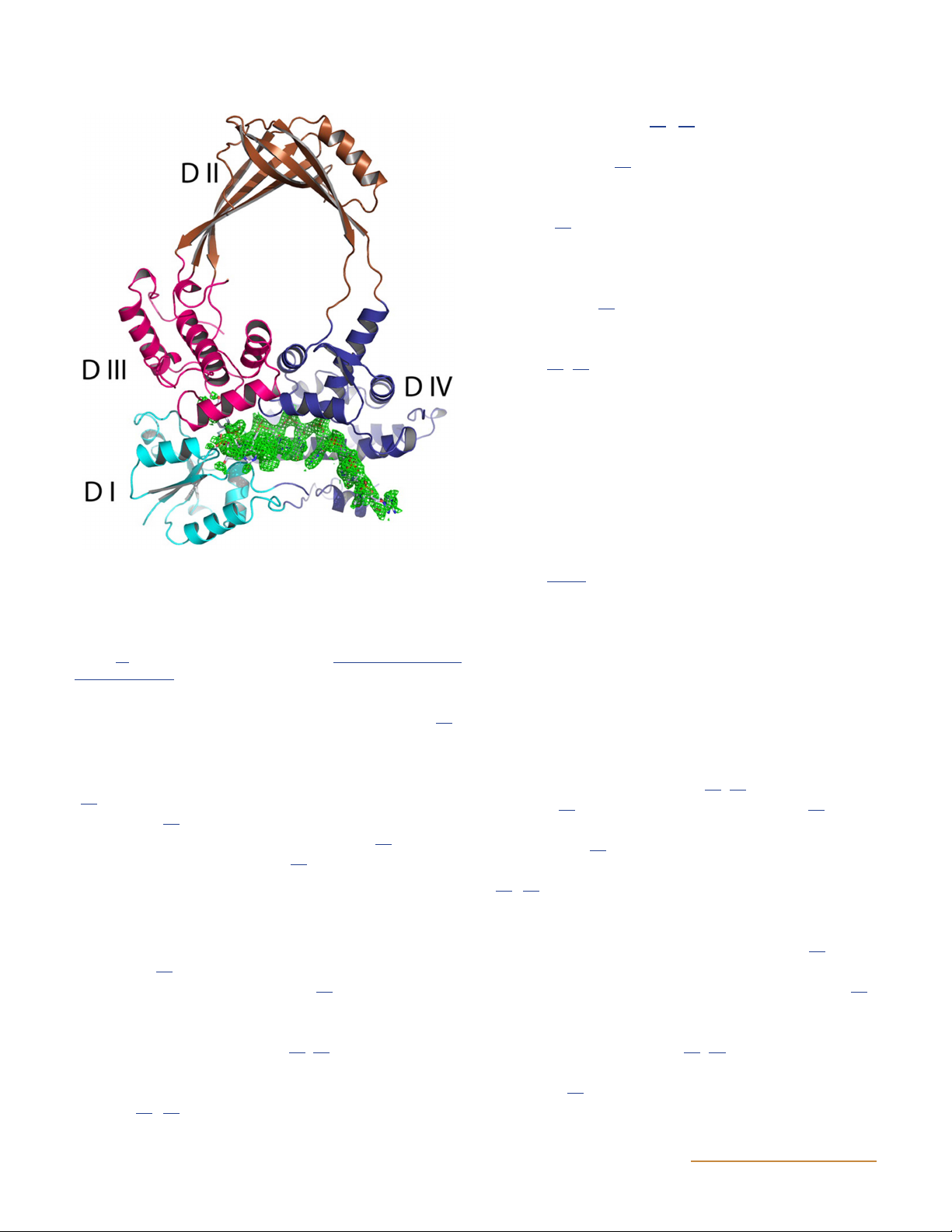
Figure 7 Structure of an N-terminal fragment of E. coli DNA topo-
larities (Fig. 6). Although the main topic of this review
isomerase I in a covalent complex with DNA. A ribbon representa-
is prokaryotic enzymes, it is useful to summarize what we
tion of the overall structure of the protein is presented, with four
know about the eukaryotic enzymes first so that com-
subdomains (DI to DIV) shown in different colors. The bound DNA
is shown in green as an electron density map. (Reprinted from ref- parisons can be made.
erence 23 with permission of the publisher.) doi:10.1128/ecosalplus. ESP-0010-2014.f7 Topo II
Eukaryotic topo II is a type IIA topoisomerase that can
in the hyperthermophile Methanopyrus kandleri (48).
relax both positively and negatively supercoiled DNA in
Topo V relaxes supercoiled DNA by a controlled rota-
an ATP- and Mg2+-dependent manner. It can also cate-
tion/swivel mechanism by nicking one strand on the
nate and decatenate DNA and has been found in many
DNA and allowing the other strand to rotate around it
eukaryotes, including humans (64, 65), Drosophila mela-
(51). It has been shown to relax around 12 turns of DNA
nogaster (66), and Saccharomyces cerevisiae (19). Most
per second (51), as opposed to type IB topoisomerases
higher eukaryotes contain two isoforms, termed topo IIα
which relax 19 turns of DNA per second (52). Topo V
and topo IIβ (67), which appear to be expressed at dif-
also has a role in DNA repair (53).
ferent times in the cell cycle and in different cell types
(68, 69). Topo IIα is found in proliferating cell types, Reverse gyrase
and expression peaks during the G and M phases of the 2
Reverse gyrase is a type IA topoisomerase that is found
cell cycle. Topo IIβ is found in all cell types, and its ex-
in thermophilic and hyperthermophilic archaea and
pression is constant throughout the cell cycle (70). More
eubacteria (54). It was initially discovered in the acido-
recently, it has been found that Topo IIβ might play a
thermophilic archaeon Sulfolobus (55). The enzyme can
role in cell differentiation and tissue development (71).
relax negatively supercoiled DNA, but interestingly, it
Topo II is required for the condensation, maintenance
can also introduce positive supercoils into relaxed DNA
of structure, and segregation of daughter chromosomes
in an ATP-dependent manner (56, 57). The crystal struc-
following DNA replication (65, 72), and it has also been
ture of reverse gyrase has been determined and reveals
linked to chromosome condensation during apoptosis in
the C-terminal domain to resemble a type 1A topoiso-
mammals (73). Many studies have also shown topo II in
merase (58, 59). The N-terminal domain consists of a
S. cerevisiae to be cell cycle regulated. 8 ASMScience.org/EcoSalPlus DNA Topoisomerases
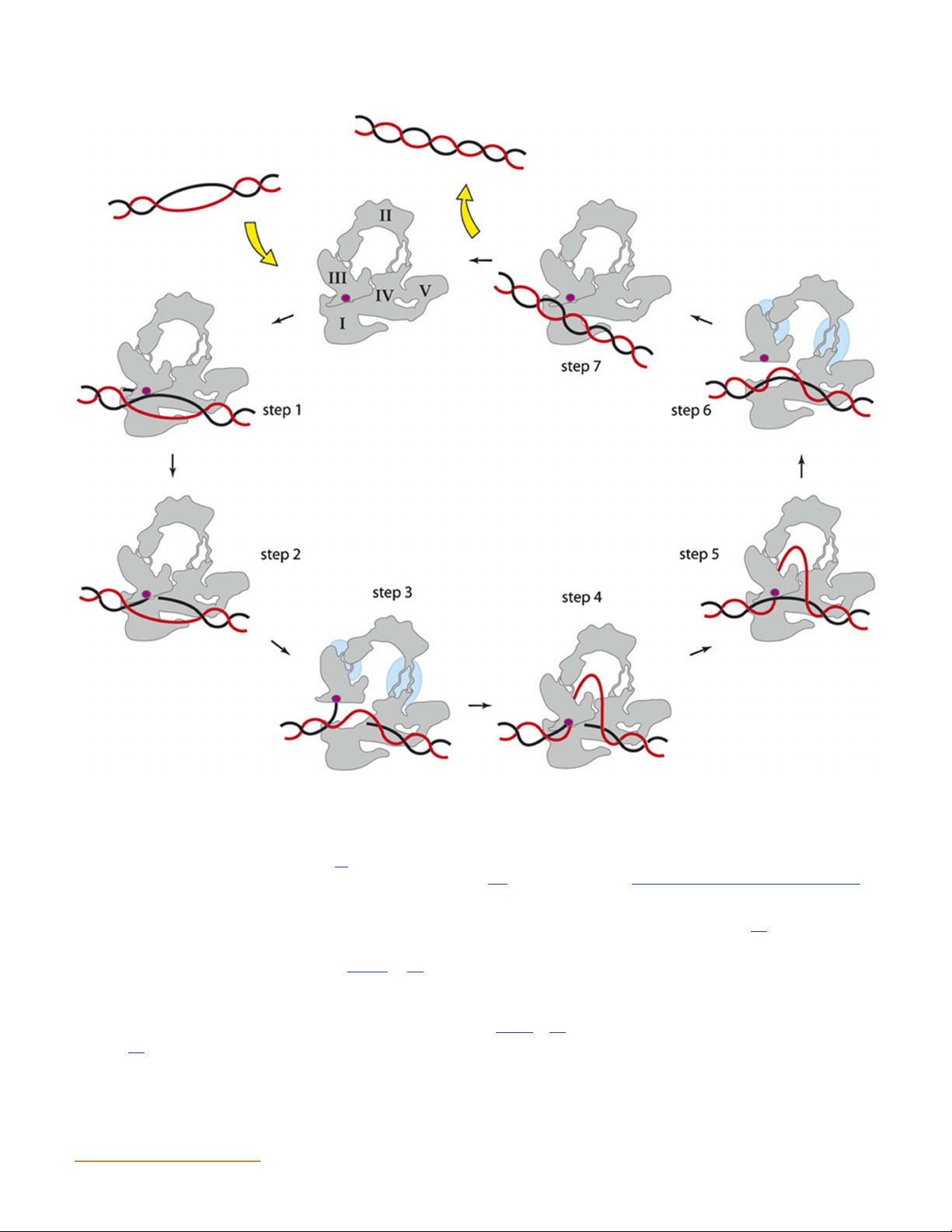
Figure 8 Proposed mechanism for E. coli topo I. The enzyme binds DNA (T segment in red, G segment in black; not to scale) and cleaves one
strand (active-site tyrosine in purple), forming a 5′-phosphodiester linkage. The complementary strand is passed through the gap and into the
central cavity of the enzyme. The light blue circles indicate areas of structural changes during the open conformation of the enzyme. The nick is
resealed, and the strand is released. It is possible that the cycle proceeds in reverse with the T segment being passed out of the enzyme rather than
in (steps 7 through 1 rather than 1 through 7) (24). (This figure was published in Viard T, de la Tour CB. 2007. Type IA topoisomerases: a simple
puzzle? Biochimie 89:456–467. Copyright © 2007 Elsevier Masson SAS. [314] All rights reserved.) doi:10.1128/ecosalplus.ESP-0010-2014.f8
Topo II has homology to DNA gyrase and topo IV (see
includes nuclear localization signals (76). The structure
below). The N-terminal domain of topo II aligns with
of residues 1 through 1177 (fully active construct) of
GyrB and the topo IV subunit ParE (Fig. 6) (74); the
S. cerevisiae topo II complexed with ADPNP (5′-adenylyl
C-terminal domain aligns with GyrA and ParC. It is
β,γ-imidodiphosphate, a nonhydrolyzable ATP analog)
thought that topo II may have evolved following the
and DNA has been determined by X-ray crystallography
fusion of the genes encoding the A and B subunits of
(Fig. 9) (77). It shows a homodimer with the N-terminal
gyrase (75). One area where topo II, topo IV, and DNA
domains in a domain-swapping conformation (i.e., the
gyrase differ is the C terminus. In DNA gyrase and topo
subunits wrap around one another). A previous structure
IV, this domain is important mechanistically, whereas
of a 92-kDa (residues 410 through 1202) fragment of
in topo II, it is thought to have a regulatory role and
yeast topo II in a complex with a DNA oligonucleotide ASMScience.org/EcoSalPlus 9 Bush et al.
Figure 9 Structure of truncated (amino acids 1 through 1177) S. cerevisiae topo II bound to DNA and ADPNP. One monomer is shaded grey, and
the other is colored by functional region. WHD is the winged-helix domain, TOPRIM is the topoisomerase-primase domain. The black box
indicates the position of ADPNP, and green indicates DNA. (Reprinted from reference 77 with permission of the publisher.) doi:10.1128/ ecosalplus.ESP-0010-2014.f9
(78) revealed that topo II introduces a 150° bend into the
the unidirectional movement of the T segment. The G
bound G segment of DNA. However, it should be noted
segment is religated; following the hydrolysis of the sec-
that the extent of the bend in the co-crystal structure may
ond ATP molecule, the T segment passes out of the en-
be influenced by crystal packing and the use of a doubly
zyme through the bottom gate, and the release of the
nicked DNA substrate. The structure of the ATPase do-
ADP molecules allows the enzyme to return to its origi-
main of yeast topo II, in a complex with ADPNP and the
nal conformation (77). Note that although this mecha-
chemotherapeutic agent ICRF-187, has also been pub-
nism is generally referred to as a “two-gate” mechanism
lished (79). This structure is similar to that of the ATPase
(83, 84), most type II topoisomerases actually possess
domain of GyrB (see below) but has some differences,
three protein interfaces (Fig. 9), the exception being the
such as a smaller central cavity (6 Å) that would appear to type IIB enzymes (see below).
be unable to accommodate a DNA duplex, in contrast to
that seen in the GyrB structure (80). Topo VI
A two-gate mechanism for topo II action, similar to that
Topo VI is an archaeal type IIB topoisomerase that is
for DNA gyrase, has been proposed (81, 82). The gate (G)
found in all known archaea but has also recently been
segment of DNA is bound and bent across the dimeric
found in plants (85, 86, 87) and in the apicomplexan
enzyme at the interface between the C-terminal (DNA-
parasite Plasmodium (88). In Arabidopsis thaliana it is
binding) domain and the N-terminal (ATPase) domain.
involved in endoreduplication (89), while in Plasmodium
The binding of ATP to the ATPase region results in the
spp. it is thought to play a role in schizogeny (88). Topo
capture of the transport (T) segment. Hydrolysis of one
VI is able to decatenate circular DNA and relax both pos-
molecule of ATP to ADP triggers cleavage of the double-
itive and negative supercoils, and it acts as an A B het- 2 2
stranded G-segment DNA, with a 4-bp stagger between
erotetramer (85). Apart from three motifs in the ATPase
the cuts in the two strands. The T segment passes through
domain and the topoisomerase-primase (TOPRIM) fold,
the gap in the G segment and into the cavity formed by the
topo VI shows no obvious sequence homology to other
two C-terminal domains. Following strand passage, the
type II topoisomerases and, therefore, is in its own sub-
two ATPase domains rotate around each other, ensuring
family (type IIB) (85, 90). The structures of the topo VI A 10 ASMScience.org/EcoSalPlus DNA Topoisomerases
subunit (topo VIA) from Methanococcus jannaschii and
organisms (e.g., Staphylococcus aureus, Oceanobacillus
topo VIB from Sulfolobus shibatae have been resolved
iheyensis, and Macrococcus caseolyticus), the correspond-
(91, 92), as well as the structures of topo VIB in a variety
ing genes are termed grlA and grlB (104, 105, 106). The
of conformations involving a range of nucleotides (91,
ParC subunit (84 kDa in E. coli) and the ParE subunit
93). More recently, however, the structure of intact topo
(70 kDa) are homologous to GyrA and GyrB, respectively
VI from Methanosarcina mazei has been determined by
(Fig. 6). However, topo IV does show some structural
using a combination of X-ray crystallography and X-ray
differences from DNA gyrase; unlike gyrase, it is unable
scattering analysis (94, 95). From these structures it is
to introduce negative supercoils into DNA (107). Topo
evident that one major difference between the A subunit
IV is also around 100 times more active at decatenation
structure of topo VI and those of other type II topo-
in vivo in E. coli cells than is DNA gyrase (11). Although
isomerases is the lack of a post-strand passage cavity (i.e.,
topo IV, and not gyrase, is responsible for decatenation in
it has only two protein interfaces, rather than the three
vivo, gyrase mutants have problems decatenating their
found in type IIA topoisomerases) (92, 94). Another dif-
chromosomes. This finding implies that DNA compac-
ference is that the double-stranded breaks made by topo
tion by gyrase is necessary for the action of topo IV (108),
VI have a 2-bp stagger in contrast to the 4-bp stagger
and, indeed, one of the roles for gyrase (see below) can
created by the type IIA topoisomerases (96). The A sub-
be viewed as supercoiling DNA catenanes to make them
unit of topo VI is homologous to a protein called Spo11,
better substrates for topo IV. Further to this, topo IV has
which is ubiquitous in eukaryotes and is involved in ini-
been shown to be processive on positively supercoiled
tiating homologous recombination during meiosis by
DNA but distributive on negatively supercoiled DNA.
cleaving DNA. In this sense, Spo11 is similar to a topo-
Positively supercoiled DNA (having left-handed cross-
isomerase that does not religate the DNA after cleavage
ings) often occurs ahead of replication forks while right-
(97). The B subunit of topo VI binds and hydrolyzes
handed crossings (negatively supercoiled DNA) are often
ATP, and the ATPase region shows structural similarity
associated with precatenanes and catenated DNA (8, 101,
to the ATPase regions of the type IIA topoisomerases,
109, 110, 111). DNA replication is stopped more quickly
despite the largely limited sequence homology. The struc-
as a result of mutations in both topo IV and gyrase than
tural work on the topo VIB subunit has revealed a de-
as a result of a mutation in gyrase alone (9). Therefore, it
tailed outline of the nucleotide hydrolysis events and the
seems that despite their sequence similarities, gyrase and
associated protein conformational changes (91, 93). It is
topo IV have quite distinct cellular roles. Topo IV has the
likely that other topo II enzymes go through a similar
predominant role in decatenation (and unknotting),
series of events. Overall, the structures of topo VI have
whereas gyrase is the only supercoiling enzyme (11, 102,
given valuable insights into the mechanism of strand pas-
108, 112). Topo IV also plays a major role in chromo-
sage by this enzyme and other topoisomerases.
some segregation after DNA replication with the help of
motor proteins and cytoskeletal components (5, 113).
Very recently a new type IIB enzyme, topo VIII, in which
the A and B subunits are fused into a single polypeptide,
The structure of a 43-kDa N-terminal fragment of ParE,
has been reported (98). Topo VIII occurs in several
in a complex with an ATP analog, has been resolved
bacterial genomes and bacterial and archaeal plasmids.
(114). This structure (Fig. 10) shows significant similarity
It is the smallest known type IIB enzyme and could be a
to that of the corresponding region of GyrB (see Fig. 11).
promising model for future structural and mechanistic
The structure of this domain gives important insight studies.
into the mechanism of ATP hydrolysis and the actions of
the aminocoumarin antibiotics, which also bind to this
region of the protein (see below). Topo IV
Topo IV is a bacterial type IIA enzyme that uses the
Structures of full-length E. coli ParC and the ParC
hydrolysis of ATP (99) to decatenate replication prod-
C-terminal domain (from Bacillus stearothermophilus)
ucts (100), relax positive and negative (although less ef-
have also been published (115, 116). The full-length ParC
ficiently) supercoils (101), and knot and unknot DNA
structure resembles those of fragments of GyrA and
(102, 103). E. coli and Salmonella Typhimurium topo IV
yeast topo II (81, 117, 118) (Fig. 10). One of the main
consists of two subunits, encoded by the parC and parE
structural differences between gyrase and topo IV is in
genes, which form a heterotetramer (99); in a few other
the C-terminal domains of GyrA and ParC. The GyrA ASMScience.org/EcoSalPlus 11 Bush et al.
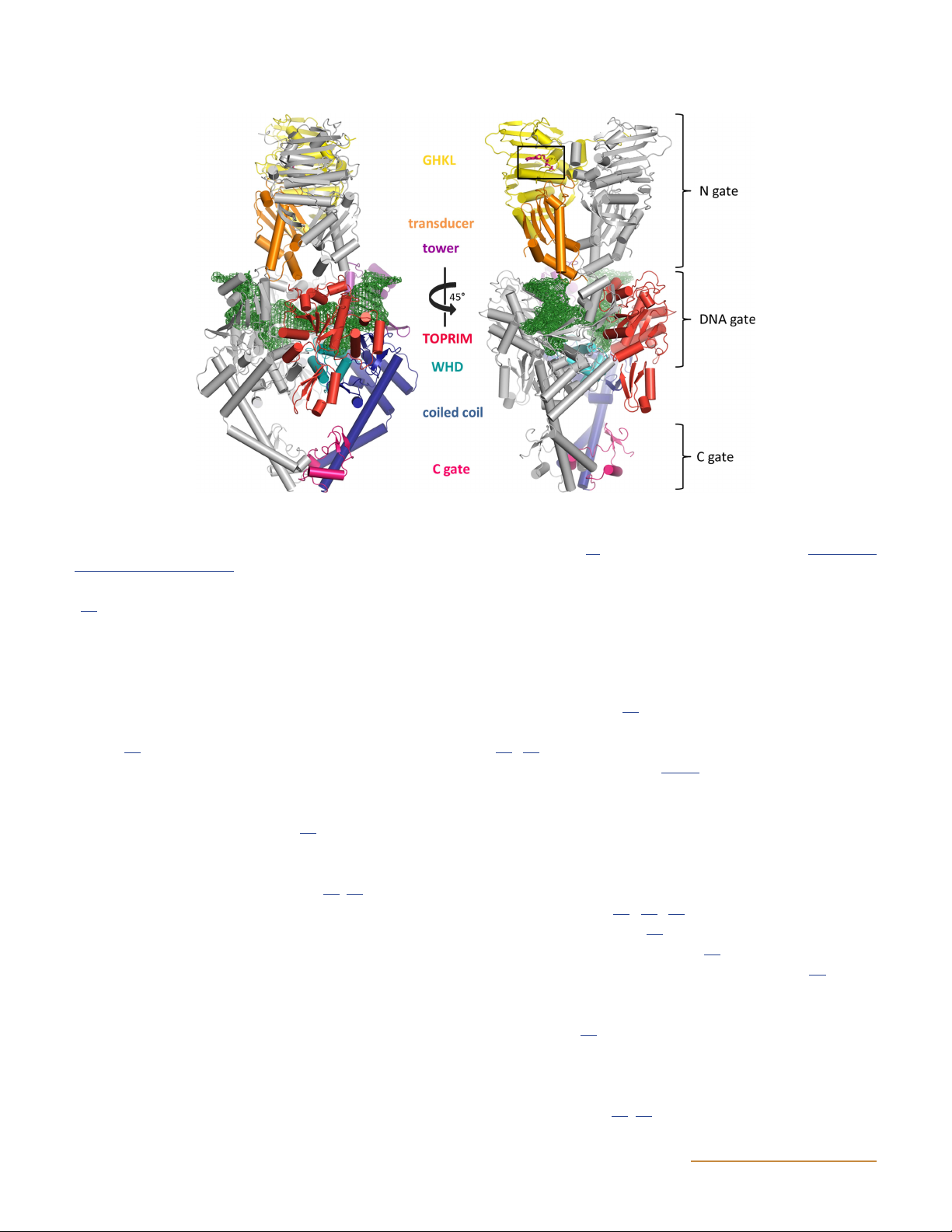
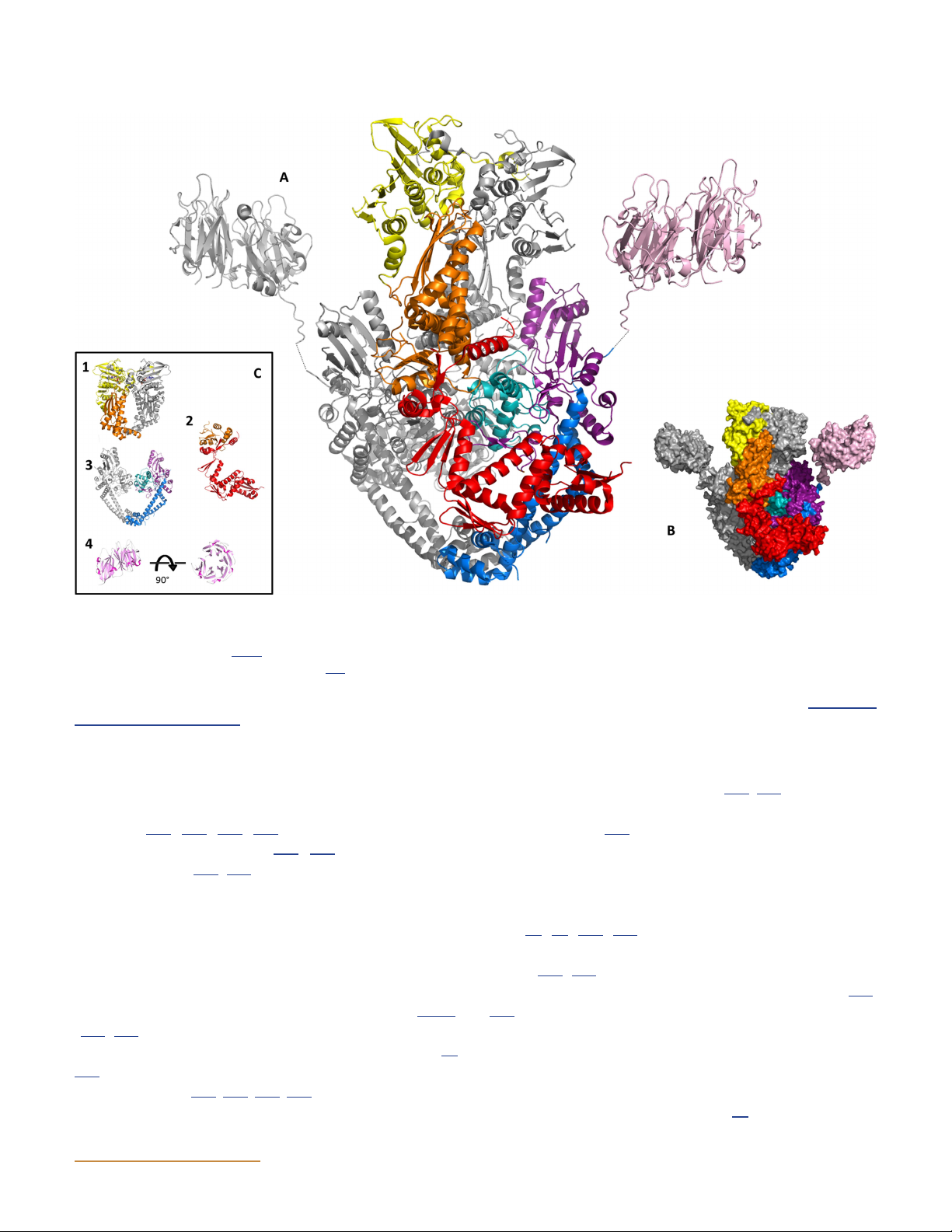
Figure 10 Structures of topoisomerase IV. (A) Structure of the ParE-ParC55 fusion construct (122) (PDB: 4I3H). Yellow indicates the GHKL
domain, orange is the transducer domain, teal is the winged-helix domain (WHD), purple is the tower domain, and blue shows the coiled-coil
domain (see Fig. 6 for domain structure). (B) Space-filled model of the structure shown in panel A. (C) ParE 43-kDa N-terminal fragment
complexed with ADPNP (black box) (PDB: 1S16) (114). It is proposed that the open conformation of ParE as seen in panel A is the conformation
pre-ATP binding whereas the conformation seen in panel C is the post-ATP-binding conformation. (D) ParC C-terminal domain in two
orientations (PDB: 1ZVT) (115). doi:10.1128/ecosalplus.ESP-0010-2014.f10
C-terminal domain forms a six-bladed β-pinwheel (see
This open conformation is thought to show the enzyme
Fig. 11) (119); the structure of the C-terminal domain
conformation prior to DNA binding.
of ParE consists of a broken five-bladed β-pinwheel
(Fig. 10) (115). The C-terminal domain of topo IV is
anchored to the N-terminal domain, which would appear DNA gyrase
to allow only minimal movement of the domain. In con-
DNA gyrase is a type IIA topoisomerase that is unique
trast, the C-terminal domain of DNA gyrase is connected
in its ability to introduce negative supercoils into cova-
to the N-terminal domain by a flexible linker, allowing
lently closed double-stranded DNA in the presence of
movement (119, 120). This distinction means that topo
ATP (123). It also uses ATP hydrolysis to relax positively
IV cannot wrap DNA in the same way as DNA gyrase
supercoiled DNA in a reaction equivalent to the intro-
can, providing an explanation for the inability of topo IV
duction of negative supercoils, despite this process being
to negatively supercoil DNA. Indeed, the deletion of the
energetically favorable (124, 125). It has also been shown
wrapping domain of gyrase converts it into a topo IV-like
to be capable of decatenation and unknotting reactions
enzyme (121). More recently, a ParE-ParC55 structure
in the presence of ATP; it is also presumably capable of
of Streptococcus pneumoniae topo IV has been resolved
catenation and knotting reactions (19, 126, 127, 128). Fur-
(122). This structure, which consists of a fusion of the
thermore, DNA gyrase can relax negatively supercoiled
full-length ParE subunits and the N-terminal domain of
DNA in an ATP-independent reaction (129, 130). DNA
ParC (ParC55) subunits, shows the ParE ATPase do-
gyrases are ubiquitous in bacteria; however, E. coli DNA
mains lying back in an open conformation and linked
gyrase has been the most intensively studied. E. coli
to the TOPRIM domain by a flexible joint (Fig. 10).
DNA gyrase is made up of two 97-kDa GyrA subunits 12 ASMScience.org/EcoSalPlus DNA Topoisomerases
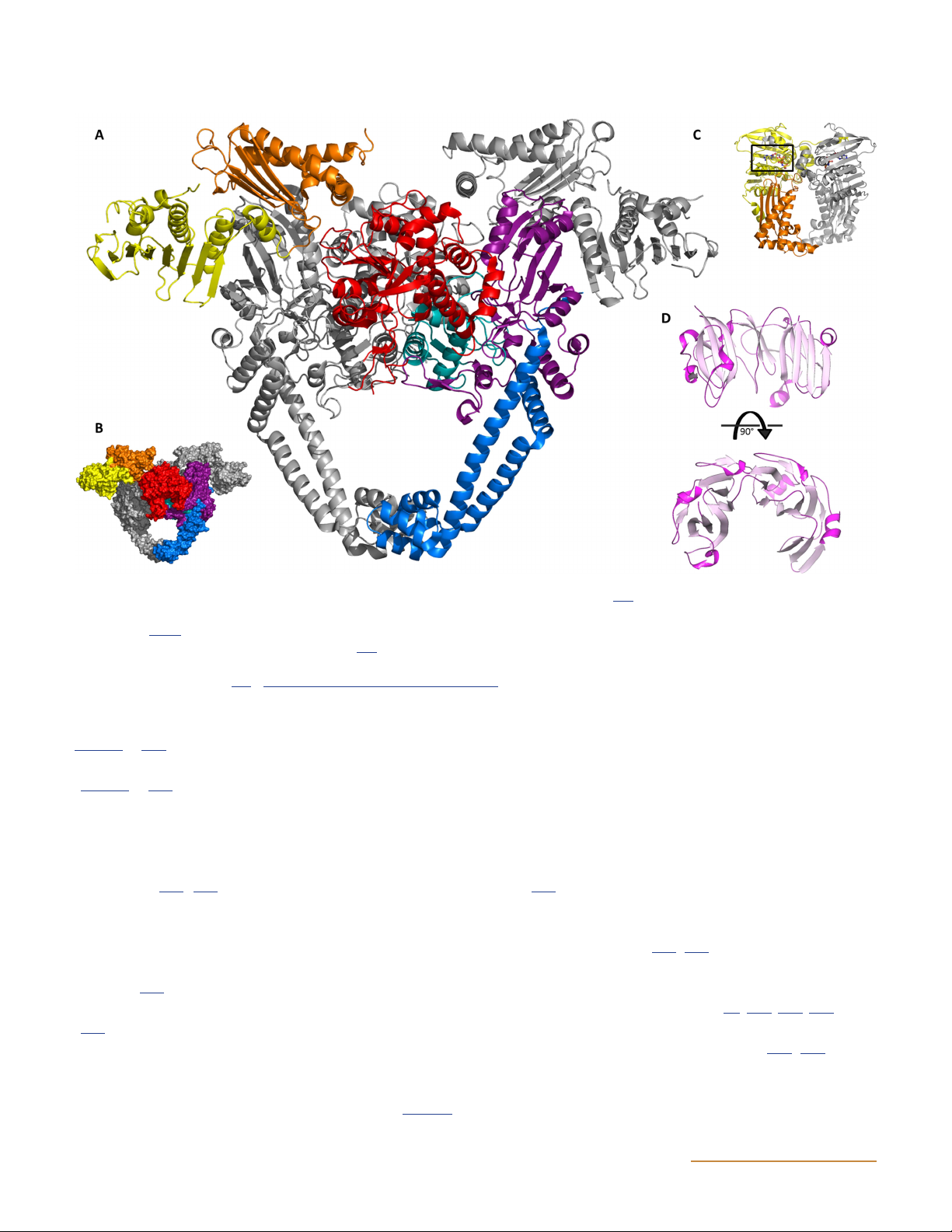
Figure 11 Structures of DNA gyrase. (A) Model of the full-length structure of DNA gyrase. Yellow indicates the GHKL domain, orange is the
transducer domain, teal is the winged-helix domain (WHD), purple is the tower domain, blue shows the coiled-coil domain, and pink indicates
the C-terminal domain (see Fig. 6 for the domain structure). The full-length protein structure was modeled on the GyrB 43-kDa fragment (PDB:
1EI1), a B-A fusion construct (PDB: 3NUH) (144), and the GyrA 35-kDa C-terminal domain (PDB: 3L6V). (B) Space-filled model of the structure
shown in panel A. (C) Four principal domains of gyrase. 1 is the E. coli GyrB 43-kDa fragment complexed with ADPNP; 2 is the E. coli GyrB
TOPRIM domain; 3 is the E. coli GyrA 59-kDa subunit; 4 is the E. coli GyrA C-terminal domain in two orientations (PDB: 1ZI0). doi:10.1128/ ecosalplus.ESP-0010-2014.f11
and two 90-kDa GyrB subunits encoded by the gyrA and
the TOPRIM domain and the tail (145, 146). E. coli GyrA
gyrB genes, respectively, and organized as an A B hetero-
consists of a 59-kDa N-terminal domain responsible for 2 2
tetramer (131, 132, 133, 134). DNA gyrases have also
DNA breakage (147) and a 35-kDa C-terminal domain
been discovered in plants (135, 136) and in apicom-
that wraps DNA. The 59-kDa domain can be further di-
plexan parasites (137, 138) but do not appear to be pres-
vided into the tower/shoulder, winged-helix, and coiled-
ent in other eukaryotes. This, along with the fact that
coil domains in line with other type IIA topoisomerases
DNA gyrase is an essential bacterial enzyme, has made
(i.e., the C-terminal domain in topo II and ParC in topo
it a successful target for several antibacterial agents.
IV) (78, 81, 118, 148). The 35-kDa domain is essential
for the ability of DNA gyrase to negatively supercoil Structure of DNA gyrase
DNA (121, 149), and its deletion converts gyrase into a
The GyrA and GyrB subunits each consist of two prin-
conventional (DNA-relaxing) enzyme like topo IV (121,
cipal domains, as revealed by limited proteolysis (Fig. 6) 150).
(139, 140). E. coli GyrB comprises a 43-kDa N-terminal
domain responsible for ATP binding and hydrolysis (80,
The protein structures of all the gyrase domains have been
141) and a 47-kDa C-terminal domain that interacts with
resolved. The first gyrase domain structure to be deter-
GyrA and DNA (141, 142, 143, 144). The 47-kDa domain
mined was that of the E. coli GyrB 43-kDa N-terminal
of GyrB may be further subdivided into two subdomains,
domain in a complex with ADPNP (80). The structure is a ASMScience.org/EcoSalPlus 13 Bush et al.
dimer (Fig. 11); each monomer consists of an N-terminal
(Fig. 11) in which each β-strand backs against and com-
ATP-binding site (amino acids 2 to 220) and a C-terminal
pletes the previous blade of the structure. The outer
portion that forms the walls of a central ∼20-Å cavity,
two-thirds of the surface of the structure is basic in
potentially large enough to hold a DNA duplex (151).
charge, suggesting that this region may be involved in the
The N-terminal portion contains the residues involved
binding and bending of DNA. Although they all share
in dimer contacts (amino acids 2 to 15) and also four
this basic structure, they have slight structural differ-
motifs conserved in members of the GHKL ATPase/
ences; e.g., the E. coli, X. campestris, and M. tuberculosis
kinase superfamily of proteins (152). Two residues in the
GyrA C-terminal domains have a spiral shape, unlike
C-terminal portion (Q335 and K337) have been shown to
the B. burgdorferi structure, which is flat. Another im-
interact with ATP (153). The central cavity formed by the
portant structural feature of the C-terminal domain is the
C-terminal portion is lined with positively charged argi-
7-amino-acid motif called the GyrA-box (158, 159). This
nine residues. Mutagenesis studies revealed that at least
motif is crucial to supercoiling activity and is found on a
one of these residues is important for DNA binding and
loop between blades 1 and 6 (158, 160). strand passage (151).
No high-resolution structures of the whole DNA gyrase
The structure of the GyrB C-terminal domain has been
enzyme have been presented to date. However, low-
solved from E. coli and Mycobacterium tuberculosis DNA
resolution structures of the entire GyrA protein and the
gyrase (144, 146, 154). These structures were also shown to
GyrB protein have been determined using small-angle
be dimers forming a “crab-like” structure with the globular
X-ray scattering (SAXS) (120, 145, 161). A GyrB-A fusion
TOPRIM domains forming the “body” and the tail do-
structure has also been elucidated by supramolecular
mains extending out to appear “claw-like.” The domains
mass spectrometry and 3D cryoelectron microscopy
are linked by a loop-helix-loop region. The TOPRIM do-
(cryo-EM) (162). Recently, a number of GyrB-A fusion
main contains three acidic residues (E459, D532, and
structures with DNA and a quinolone drug bound have
D534) responsible for binding the magnesium ion neces-
been resolved by X-ray crystallography (see below), as
sary for the cleavage-religation reaction. These residues
well as structures with topo IV, DNA, and drugs (163,
are highly conserved among DNA gyrases (144, 146, 164, 165).
154). The E. coli GyrB C-terminal domain differs from
the M. tuberculosis structure by a 170-amino-acid insert
Ab initio modeling shows the 59-kDa N-terminal domain
which adopts an extended fold. This lies alongside the
forming a dimeric core, with a pear-shaped density pat-
coiled-coil domain of the GyrA subunit (Fig. 11). This
tern on either side. These densities may accommodate
extra insert is thought to have a role in DNA binding
the crystal structure of the GyrA C-terminal domain (119)
and DNA-stimulated ATPase activity (142, 144).
attached to the N-terminal domain by a flexible linker.
The cryo-EM structures complexed with ADPNP, cip-
The structure of the 59-kDa N-terminal domain of E. coli
rofloxacin, and DNA indicate that the GyrA C-terminal
GyrA was resolved in 1997 (118). This structure is also a
domains are elevated, alongside the DNA gate and GyrB
dimer and contains a 30-Å central cavity (Fig. 11). There
subunits (Fig. 12) (162). The molecular envelope of GyrB
are two dimer interfaces, at the top and bottom of the
has a “tadpole” shape, with the ATPase domain structure
structure, which comprise the DNA gate and the C-gate,
of GyrB (80) fitting into the head of this envelope
respectively. The interface at the top of the dimer con-
and the remainder being made up of the TOPRIM fold
tains the active-site tyrosines, which form phospho-
(81) and the tail subdomains. Investigation by analytical
tyrosine bonds with the 5′ ends of the broken DNA. The
ultracentrifugation has revealed that GyrB, unlike GyrA,
region across the dimer interface at the top of the domain
is predominantly a monomer in solution (120, 145). The
provides a positively charged saddle, which is proposed
structural information from topo II structures (81) im- to promote DNA binding.
plies that GyrB sits above GyrA in the complex (Fig. 9).
This is corroborated by the SAXS and cryo-EM data,
The structure of the 35-kDa C-terminal domain of DNA
which imply that the GyrB ATPase domains are posi-
gyrase from a number of bacterial species, including
tioned above the DNA cleavage active site at an angle
Borrelia burgdorferi, E. coli, M. tuberculosis, and Xan-
between 60 and 105° (161, 162). The data also suggest
thomonas campestris, has been determined (119, 155,
the ATPase domain is angled 15 to 20° toward one of
156, 157). This domain forms a six-bladed β-pinwheel
the GyrA C-terminal domains (162). 14 ASMScience.org/EcoSalPlus DNA Topoisomerases
Figure 12 Cryo-EM map of the DNA-bound complex modeled with the crystal structures of DNA gyrase domains alone (A) and with duplex DNA
(B). In particular, the crystal structures of the ATPase (PDB:1EI1) and the DNA-binding-cleavage domain in the presence of ciprofloxacin
(PDB:2XCT) were modeled into the core of the map with the two additional densities on both side of the core enzyme accommodating the
C-terminal domains (PDB:3L6V). (Reprinted from reference 162 with permission of the publisher.) doi:10.1128/ecosalplus.ESP-0010-2014.f12
Mechanism of action of DNA gyrase
of ATP; however, ATP (or ADPNP) is crucial for strand
Biochemical characterization of the roles of GyrA and
passage to occur (174). Following DNA binding and
GyrB has revealed details of the mechanism of super-
wrapping, ATP is bound to the N-terminal domains of
coiling by gyrase. More recently, single-molecule ex-
the two GyrB subunits, resulting in their dimerization
periments have corroborated this biochemical data and
and the closure of the clamp. This closure traps the T
further extended our knowledge and understanding of
segment in the complex (174, 175). In E. coli a small this mechanism.
acidic tail on the GyrA C-terminal domain has been im-
plicated in the coupling of ATP to DNA wrapping, thus
Negative supercoiling occurs via a two-gate mechanism
controlling supercoiling (176).
(128) (Fig. 13). A section of DNA termed the gate or G
segment binds across the top dimer interface of the GyrA
The active-site tyrosines form phosphotyrosine bonds
N-terminal domains (118, 133). Upon binding, the G seg-
with the G segment, generating a double-strand break
ment is bent at an angle of about 70°, which is much less
with 4-bp overhangs (147, 171). Two Mg2+ ions bound
than in the S. cerevisiae topo II structure (78, 162).
within the TOPRIM fold of GyrB are required for the
Binding of the G segment induces an upward movement
cleavage of the DNA strands (143). The top dimer in-
of the GyrA C-terminal domains, resulting in an adjacent
terface is pulled apart, and with it the G segment, allow-
section of the DNA becoming wrapped around the
ing the T segment to pass through into the cavity formed
C-terminal domain (166, 167). This wrapping positions a
by the GyrA N-terminal domains. The GyrA cavity car-
further DNA section, termed the transport or T segment,
ries a positive charge and so provides a favorable envi-
across the G segment at an angle of about 60° (162, 166). ronment for DNA (118).
The GyrA box is thought to ensure the orientation of the
T segment in a way that favors DNA supercoiling (158,
The G segment is religated, and the T segment is released
159, 160). This wrapping by the GyrA C-terminal do-
through the bottom gate of the GyrA N-terminal do-
main provides gyrase with its unique ability to supercoil
mains. It is not currently clear what drives the movement
DNA (119, 149, 156). The total length of DNA bound by
of the T segment at this stage. It has been proposed that
gyrase is estimated to be between 120 and 150 bp (168,
it may be the closure of the top gate, which makes the
169, 170, 171, 172). DNA binding has also been shown
GyrA cavity smaller (177). More recently it was proposed
to induce narrowing of the GyrB N-terminal domains
that the closing and swiveling observed in the GyrB
(173). DNA wrapping and the presentation of the T
subunits may ensure unidirectional movement of the
segment has been demonstrated to occur in the absence
T segment (162, 173). The rotating of the N-terminal ASMScience.org/EcoSalPlus 15 Bush et al.
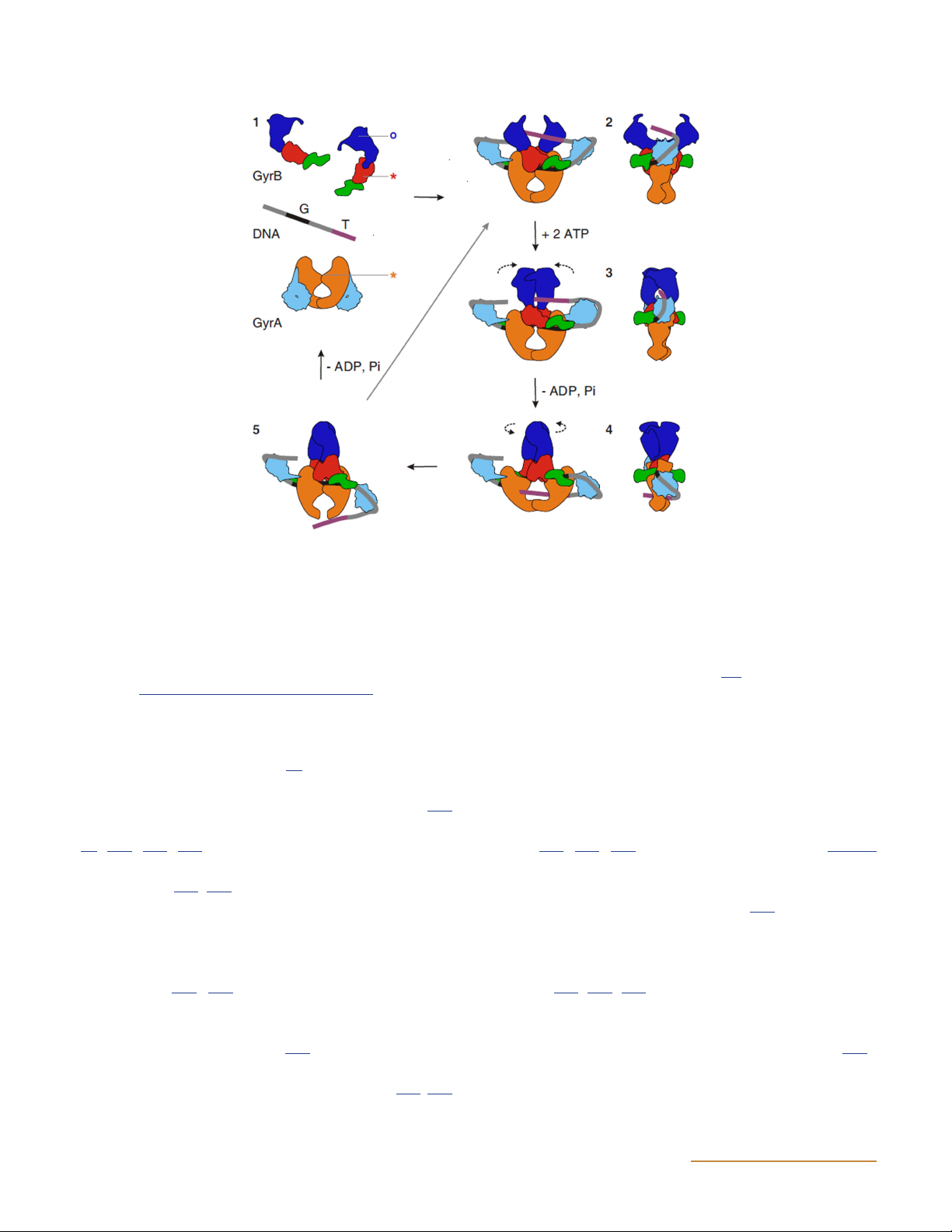
Figure 13 Model for negative supercoiling by DNA gyrase. The domains are colored as follows: GyrB43, dark blue; GyrB TOPRIM, red; GyrB tail,
green; GyrA59, orange; GyrA C-terminal domain, light blue. The G and T DNA segments are colored black and purple, respectively. 1, subunits
and DNA in their proposed free states in solution. Stars indicate the active-site residues for DNA cleavage, and the circle indicates the ATP-
binding pocket. 2, The G segment binds across GyrA at the dimer interface, and the GyrA C-terminal domain wraps the DNA to present the
T segment in a positive crossover. 3, ATP is bound, which closes the GyrB clamp capturing the T segment, and the G segment is transiently
cleaved. 4, Hydrolysis of one ATP molecule allows GyrB to rotate, the DNA gate to widen, and the T segment to be transported through the
cleaved G segment. 5, The T segment exits through the C gate, and the G segment is religated. The hydrolysis of the remaining ATP molecule
resets the enzyme. The right panel shows the side view for illustrations 2 through 4. (Reprinted from reference 145 with permission of the
publisher.) doi:10.1128/ecosalplus.ESP-0010-2014.f13
domains (GyrB equivalent) in S. cerevisiae has also been
has received significant attention, and it seems that
shown in its crystal structure (77).
gyrase-catalyzed ATPase activity is not tightly coupled
to supercoiling; as gyrase goes through a supercoiling
ATP hydrolysis allows the resetting of the enzyme (125);
reaction, ATP continues to be hydrolyzed at a high rate,
ADPNP is sufficient for the capture of the T segment
even after the enzyme has reached the supercoiling end-
(83, 178, 179, 180) and for a single strand-passage re-
point (125, 182, 183). It should be noted that Fig. 13
action to occur, but the enzyme is then trapped in an
suggests that the two ATPs may be hydrolyzed sequen-
inactive state (125, 181). More detailed studies of the in-
tially in the supercoiling mechanism; this is based on
teraction of ADPNP with gyrase have provided evidence
work carried out with yeast topo II (186) and may not
of the cooperative role of the two ATP-binding sites in
necessarily apply to gyrase. Indeed, work on gyrase sug-
the supercoiling cycle and examined the coupling of nu-
gests that ATP binding may be sufficient to carry out a
cleotide binding to strand passage at different levels of
complete strand passage reaction (ΔLk – 2) without hy-
supercoiling (182, 183). To date, the mechanism that
drolysis (125, 182, 183), which may be required only to
drives ATP hydrolysis is uncertain; however, the ATPase
reset the enzyme. However, it is also worth noting that
activity has been revealed to be stimulated by cleaved
gyrase can carry out limited catalytic supercoiling with
DNA in the presence of GyrA (184). It has been proposed
the binding and hydrolysis of only one ATP (187).
that the rate-limiting aspect of the DNA supercoiling re-
The full intricacies of the gyrase supercoiling reaction
action is the rate of ADP and phosphate release (178, 185).
and the coupling of ATP binding/hydrolysis are still
The coupling of supercoiling to ATP binding/hydrolysis under active investigation. 16 ASMScience.org/EcoSalPlus DNA Topoisomerases
DNA gyrase can also relax negatively supercoiled DNA,
More recently 3,4-dimethoxyphenyl bis-benzimidazole
which occurs as the reverse of the reaction described
has been shown to target E. coli topo I and to have low
previously, with the T segment passing through the en-
MICs for a range of E. coli strains (189). Taken together,
zyme in the opposite direction (188). This reaction is
it seems likely that clinically relevant compounds that
ATP independent, since it is energetically favorable, and
target bacterial topo I will be available in the future.
is far less efficient than the supercoiling reaction (129,
130). DNA gyrase can also relax positively supercoiled DNA Gyrase
DNA. This reaction occurs in the same way as negative
At the moment the only bacterial topoisomerase target
supercoiling and requires ATP, even though it is ener-
that is commercially significant is DNA gyrase, although
getically favorable (141). The catenation-decatenation
a number of gyrase-targeting agents also target the sister
and knotting-unknotting reactions performed by gyrase
enzyme topo IV. There are two well-known classes of
are also ATP dependent (19, 126).
drugs that target gyrase: the aminocoumarins and quino-
lones (190). The quinolones are synthetic, whereas the
DRUGS AND TOXINS THAT TARGET BACTERIAL
aminocoumarins are products of Streptomyces species DNA TOPOISOMERASES (Table 2).
DNA topoisomerases are important targets for antimi- Aminocoumarins
crobial drugs (31). DNA gyrase is essential for the sur-
vival of bacteria but is largely absent in eukaryotes and
The aminocoumarins are more potent inhibitors of
is therefore an ideal drug target. DNA topoisomerase I is
gyrase than are the quinolones in vitro, but their low
regarded as nonessential, but the fact that its mechanism
solubility and toxicity in eukaryotes make them less use-
involves the formation of a cleavage complex with DNA
ful clinically (190). These compounds are produced
raises the possibility of exploiting this target. At the time
by Streptomyces species and include novobiocin, cloro-
of writing, there are no commercially produced antibac-
biocin, and coumermycin A (Fig. 14) (191, 192, 193, 194, 1
terial agents that target topo I, but there is ongoing work 195, 196, 197).
to find such agents. Topo I and gyrase are discussed separately.
Aminocoumarins inhibit supercoiling, leading to the
identification of DNA gyrase as the target (124). However,
the aminocoumarins do not inhibit ATP-independent Topo I
relaxation (198, 199), consistent with their being competi-
Although bacterial topo I appears to be nonessential,
tive inhibitors of ATP hydrolysis. This conclusion is sup-
the discovery of compounds that stabilize the cleavage
ported by work showing that novobiocin strongly inhibits
complex and show antibacterial activity raises the pos-
the gyrase ATPase reaction, which is relatively unaffected
sibility of new antibacterials targeted to topo I in the
by the quinolone oxolinic acid (200). Aminocoumarin-
future (30). Proof of principle for this assertion has
resistant strains of E. coli frequently contain a mutation
been provided by experiments in which a mutant form of
of Arg136 (201, 202), a residue not directly implicated in
Yersinia pestis topo I that forms a stabilized covalent
ATP binding (80). This discrepancy was explained with
complex was shown to result in cell death in E. coli (29).
the resolution of crystal structures of the N-terminal
Subsequently it was shown that compounds that enhance
24-kDa subdomain of GyrB in complexes with novobio-
DNA cleavage by topo I have antibacterial activity (32).
cin and clorobiocin (203, 204, 205); the structure of the
Table 2 Inhibitors of DNA gyrase Drug(s) or toxin Source Mode of action
Aminocoumarins (e.g., novobiocin) Streptomyces species
Competitive inhibition of ATP binding
Quinolones (e.g., ciprofloxacin) Synthetic
Stabilization of DNA cleavage complex Albicidin Xanthomonas albilineans
Stabilization of DNA cleavage complex
Simocyclinones (e.g., simocyclinone D8) Streptomyces antibioticus Prevention of DNA binding MccB17 Escherichia coli
Stabilization of DNA cleavage complex CcdB Escherichia coli
Stabilization of DNA cleavage complex ASMScience.org/EcoSalPlus 17 Bush et al.
Figure 14 Structures of aminocoumarins. doi:10.1128/ecosalplus.ESP-0010-2014.f14
corresponding region of E. coli ParE (topo IV) in a com-
and the clorobiocin and coumermycin A gene clusters 1
plex with novobiocin has also been determined (114). In-
also encode a ParY subunit, encoding a drug-resistant
deed, topo IV is a secondary target of novobiocin (206).
subunit (ParE) of topo IV (207). Differences among
There is only a partial overlap between the aminocoumarin
the gene clusters for these compounds correspond to
drugs and ATP-binding sites, with the novobiose sugar
differences in the compound structures; for example,
of novobiocin overlapping with the adenine ring of ATP
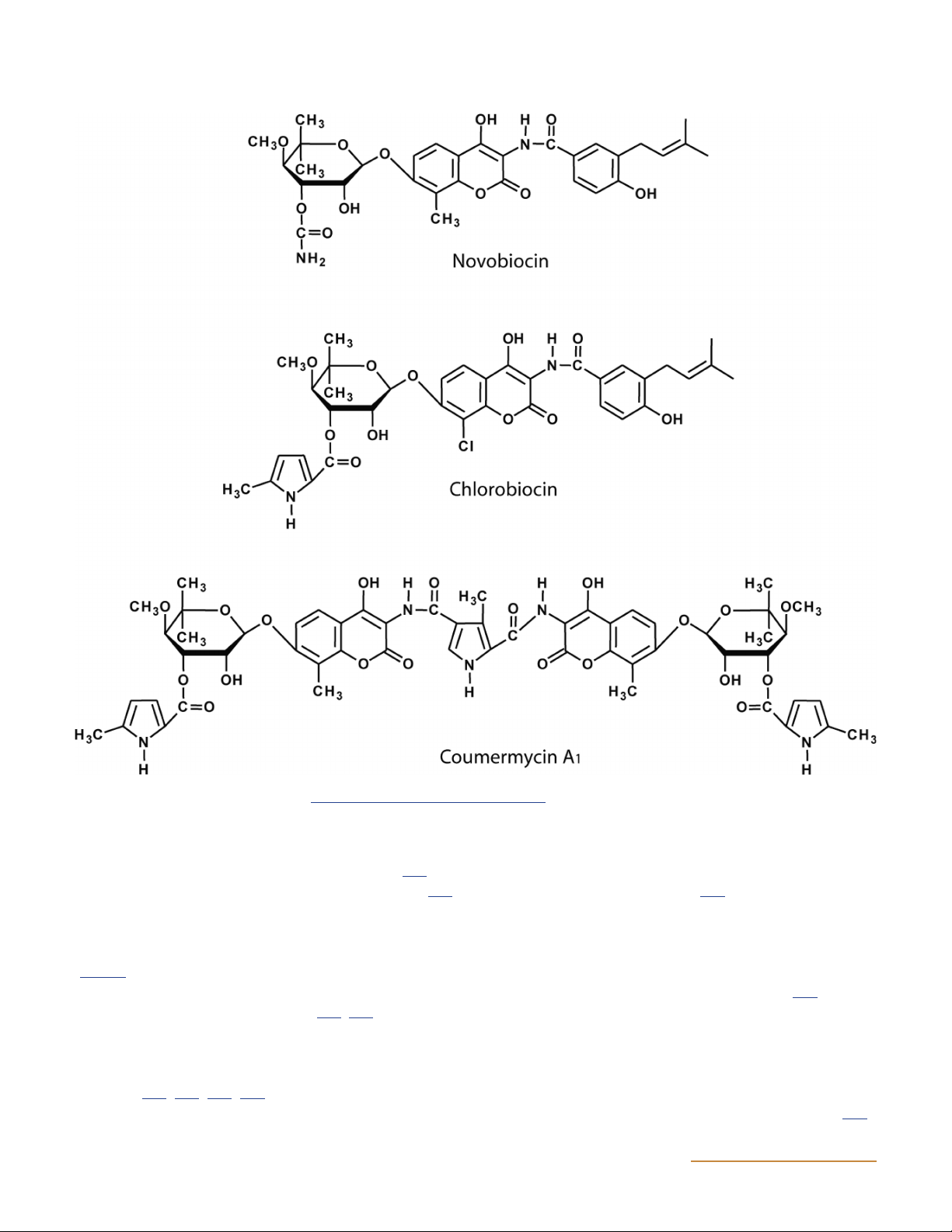
novobiocin contains a methyl group at position 8 on the
(Fig. 15). Novobiocin forms a hydrogen bond with Arg136
aminocoumarin ring, and its biosynthetic gene cluster
and has been shown to prevent the dimerization of the
contains novO, a C-methyltransferase gene (207). Cloro-
43-kDa GyrB N-terminal domains (178, 204).
biocin contains a chlorine atom at position 8 on the ring,
and the corresponding gene (clo-hal) in the clorobiocin
The biosynthetic gene clusters for novobiocin, coumer-
gene cluster has similarity to a reduced flavin adenine
mycin A , and clorobiocin have been cloned and se-
dinucleotide-dependent halogenase gene. The replace- 1
quenced (207, 208, 209, 210). All three clusters contain a
ment of the clo-hal gene in the clorobiocin gene cluster
gene encoding an aminocoumarin-resistant GyrB subunit,
with novO results in an 8′-methylated derivative (211). 18 ASMScience.org/EcoSalPlus DNA Topoisomerases
Simocyclinone D8 (SD8) has been shown to be a potent
inhibitor of both the supercoiling and relaxation activi-
ties of gyrase in vitro (218). Despite the similarities be-
tween SD8 and the classical aminocoumarins, they show
key differences in their modes of action. SD8 does not
competitively inhibit the ATPase reaction, nor does it
stabilize the gyrase-DNA cleavage complex like the quino-
lones (218); instead, it acts at an early stage in the gyrase
catalytic cycle to prevent DNA binding (218, 219). The
first crystal structure of SD8 bound to the N-terminal
domain of E. coli GyrA showed that both ends of the
molecule interact with the protein, leading to its being
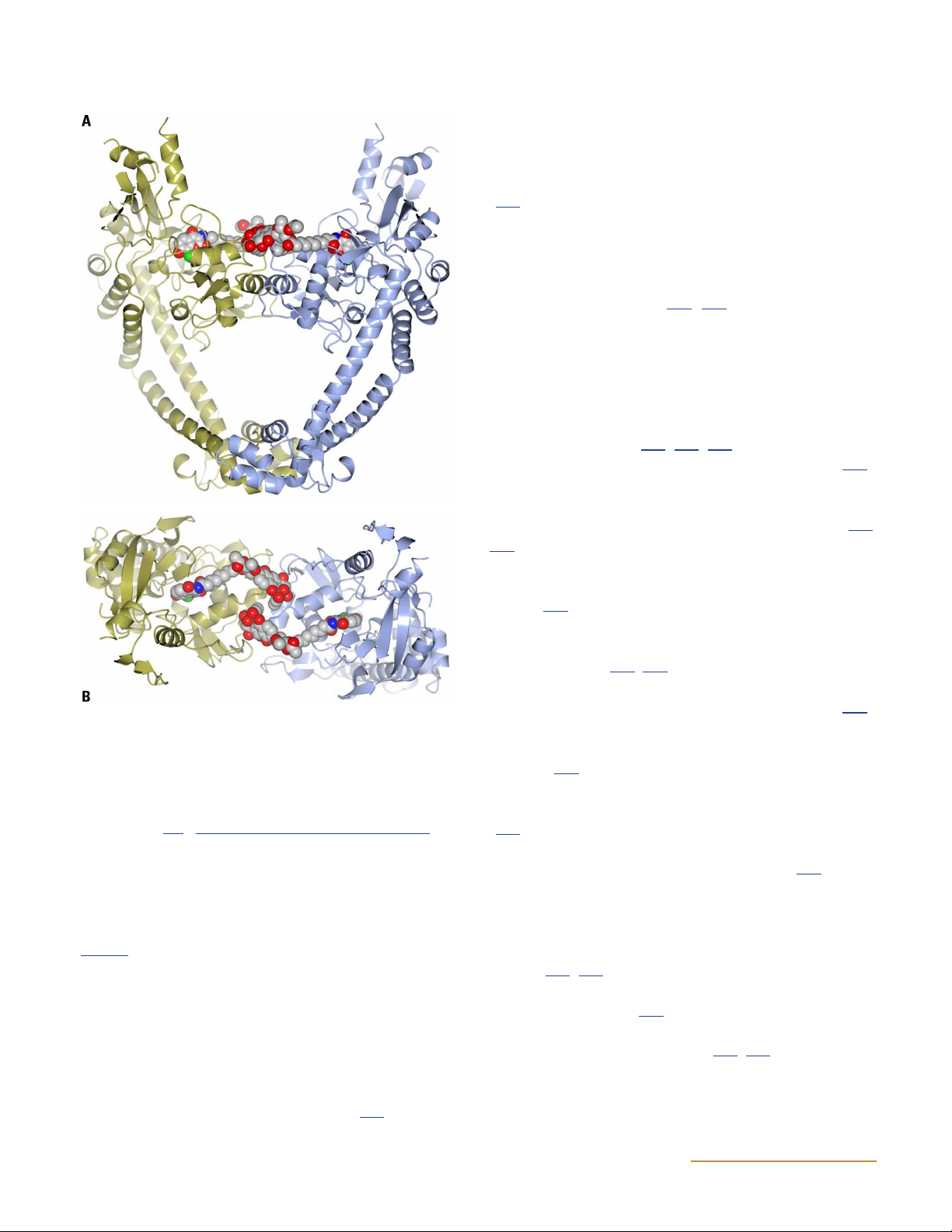
dubbed a “double-headed antibiotic” (219). This crystal
Figure 15 The binding sites of novobiocin and ADPNP in GyrB par-
structure has been more recently refined by mass spec-
tially overlap. Part of the N-terminal GyrB structure is shown, with
trometry studies (220) and subsequent X-ray crystallog-
ADPNP in red and novobiocin in blue (204). doi:10.1128/ecosalplus.
raphy (221), showing that a single SD8 molecule can ESP-0010-2014.f15
bridge both binding pockets in a single GyrA protomer (Fig. 17).
Modification of the aminocoumarin gene clusters by ge-
netic engineering therefore has the potential to generate
Although SD8 is an effective inhibitor of E. coli gyrase, it
novel antibiotics. For example, analogs of novobiocin and
is ineffective against gram-negative bacteria and is active
clorobiocin, termed novclobiocins, have been generated
against only certain gram-positive organisms (215, 222).
(212, 213). Although most of these analogs are less potent
Simocyclinones are not particularly promising as drug
than the parent molecules, novobiocin and clorobiocin,
candidates as a result of their lack of penetration into
some show equal or even greater potency. Moreover, many
bacteria and also on account of their low solubility and
of the analogs have improved physicochemical properties
toxicity in eukaryotes (215, 223), but it is hoped that
and present the possibility of developing superior che-
modification of the structure may circumvent these prob- motherapeutic agents.
lems. In particular, their novel mode of action suggests
that there are still further unexploited strategies for in- Simocyclinones hibiting bacterial gyrase.
Simocyclinones D4 and D8 (Fig. 16) are gyrase-inhibiting
antibiotics from Streptomyces antibioticus (214, 215). It Quinolones
was noted that some of the genes in the simocyclinone
The quinolones are the most therapeutically important
cluster have similarities to those in the aminocoumarin
class of DNA gyrase inhibitors, and they have been used
gene cluster, and the simocyclinone structure includes
to treat a wide range of infections (190, 224, 225, 226).
an aminocoumarin moiety (216, 217).
The quinolones traditionally have been divided into two
Figure 16 Structure of simocyclinone D8. doi:10.1128/ecosalplus.ESP-0010-2014.f16 ASMScience.org/EcoSalPlus 19 Bush et al.
increased spectrum seen in the newer generations of the
fluoroquinolones, there has also been an improvement in
the bioavailability of these drugs, as well as better tissue
penetration and improvements in safety and tolerability (227).
The quinolones have been shown to inhibit DNA super-
coiling and relaxation by binding to both gyrase and
DNA and stabilizing the formation of the gyrase-
DNA cleavage complex (198, 199). In recent years the
specifics of the interaction between quinolones and the
gyrase-DNA complex have been revealed by X-ray crys-
tallography (see below). The inhibition of DNA synthesis
by quinolones is due not to the inhibition of gyrase ac-
tivity per se but to the quinolone-gyrase-DNA complex
blocking the DNA replication machinery and hence
blocking cell growth (228, 229, 230). This effect is likely
to result in the bacteriostatic action of quinolones (231).
Cell death is likely to be due to DNA breaks, which form
a second step in the process and can occur by both pro-
tein synthesis-dependent and -independent routes (231,
232). DNA replication is stopped rapidly when DNA
gyrase is targeted with quinolone drugs, apparently due
to the collision of replication forks with cleaved com-
plexes (233). For example, norfloxacin has been shown
to cause stalled replication forks in vivo; however, this
inhibition cannot be the immediate cause of cell death,
as it is reversible (234, 235). Also, bacteria in which DNA
replication has been inhibited can subsequently be killed
by treatment with nalidixic acid or ciprofloxacin (236).
Figure 17 Structure of the N-terminal domain of GyrA (GyrA55)
The inhibition of DNA replication by quinolones results
complexed with simocyclinone D8. The protein dimer is shown in
in the induction of the SOS regulon in a RecBC-dependent
gold and blue (ribbon representation), and the bound simocyclinone
manner (237). One of the genes induced is an inhibitor
D8 dimer is shown in space-filling representation. (A) Side view.
of cell division, so the SOS response results in cell fila-
(B) Top view. Note that the polyketide end of each simocyclinone
molecule also binds to the other monomer across the dimer (DNA-
mentation, which may lead to the slow death of the cell
gate) interface (221). doi:10.1128/ecosalplus.ESP-0010-2014.f17
(238). The release of DNA with double-strand breaks
from several cleavage complexes may cause chromosome
fragmentation and leads to rapid cell death (239). Chro-
categories: the older acidic quinolones, such as nalidixic
mosome fragmentation may be dependent on or inde-
acid, which act against gram-negative bacteria, and the
pendent of protein synthesis. Protein synthesis-dependent
amphoteric fluoroquinolones, such as ciprofloxacin
chromosome fragmentation is inhibited by chloramphen-
(Fig. 18); strictly speaking, nalidixic acid is a naphthy-
icol, and it has been proposed that a suicide factor is in-
ridone, not a quinolone, but is usually grouped with the
volved (232, 240). There is evidence to support the idea
quinolone drugs. More recently, the quinolones have
that the RecB and RecC proteins are involved in nalidixic
been classified in terms of the evolution of their struc-
acid-induced breaks (241). The MICs of quinolones are
tures and clinical indications: narrow-spectrum drugs in-
10- to 100-fold lower (in vivo) than their 50% inhibitory
clude nalidixic acid; expanded-spectrum drugs include
concentrations (IC s) (in vitro) (198, 242). This property 50
norfloxacin and ciprofloxacin; broad-spectrum quino-
can be explained by a low concentration of the inhibi-
lones include levofloxacin and sparfloxacin; and “fourth-
tor in vivo triggering the downstream responses that
generation” drugs include trovafloxacin (227). With the
lead to cell death. In bacteria, susceptibility to quinolones 20 ASMScience.org/EcoSalPlus



