Dược học - Đại học y dược Huế
Dược học - Đại học y dược Huế được biên soạn dưới dạng file PDF cho các bạn sinh viên tham khảo, ôn tập đầy đủ kiến thức, chuẩn bị thật tốt cho các kì thi sắp tới. Mời bạn đọc đón xem!
Môn: Hóa dược 1 7 tài liệu
Trường: Trường Đại học Y dược - Đại học Huế 361 tài liệu
Tác giả:



















































































Preview text:
TRƯỜNG ĐẠI HỌC Y DƯỢC HUẾ
KHOA DƯỢC – BỘ MÔN DƯỢC LÂM SÀNG DƯỢC ĐỘNG HỌC
(Lưu hành nội bộ) Trương Viết Thành
Võ Thị Hồng Phượng Võ Thị Hà
Phan Đặng Thục Anh Huế - 11/2015 LỜI NÓI ĐẦU
Dược Lâm Sàng là một môn học mới được đưa vào giảng dạy và nghiên cứu so với các
môn học truyền thống của ngành Dược. Mục đích của Dược Lâm Sàng là tối ưu hóa việc
sử dụng thuốc cho bệnh nhân. Để học tốt Dược Lâm Sàng, sinh viên cần nắm được các
kiến thức căn bản trong đó có kiến thức về Dược Động Học. Môn học Dược Động Học sẽ
giúp sinh viên có được kiến thức căn bản về số phận của thuốc trong cơ thể bao gồm các
quá trình hấp thu, phân bố, chuyển hóa và thải trừ, đồng thời phán đoán được sự thay đổi
của các quá trình đó trong một số điều kiện mắc bệnh nhất định của bệnh nhân. Ngoài ra,
qua các bài giảng, sinh viên sẽ được tiếp cận với các phương pháp giám sát nồng độ
thuốc trong điều trị, đặc biệt với các thuốc có độc tính cao và thuốc có giới hạn hẹp giữa
liều có tác dụng tối thiểu và liều gây độc.
Bộ môn Dược Lâm Sàng đã cố gắng soạn thảo với tinh thần trách nhiệm cao Giáo trình
Dược Động Học nhằm cung cấp cho sinh viên một công cụ học tập và hy vọng đây là nền
tảng kiến thức giúp sinh viên lĩnh hội tốt hơn môn học Dược Lâm Sàng và các môn học khác sau này.
Bộ Môn Dược Lâm Sàng –Khoa Dược MỤC LỤC
CHƯƠNG I. DƯỢC ĐỘNG HỌC _________________________________________ 1
BÀI 1: HẤP THU THUỐC, SINH KHẢ DỤNG VÀ ĐƯỜNG DÙNG THUỐC __ 1
1. Khái niệm hấp thu thuốc _____________________________________________ 2
2. Cơ chế vận chuyển thuốc trong cơ thể __________________________________ 2
2.1. Vận chuyển thụ động (khuếch tán)__________________________________ 2
2.2. Vận chuyển thụ động nhờ chất mang ________________________________ 3
3. Sinh khả dụng - Tương đương sinh học - Tiền thuốc - Thuốc gốc và Thuốc biệt
dược _______________________________________________________________ 4
3.1. Sinh khả dụng __________________________________________________ 4
3.2. Tương đương sinh học ___________________________________________ 5
3.3. Tiền thuốc - Thuốc gốc và thuốc biệt dược ___________________________ 6
4. Các yếu tố ảnh hưởng đến hấp thu thuốc ________________________________ 7
4.1. Các yếu tố sinh - bệnh lý _________________________________________ 7
4.2. Các yếu tố ngoại lai _____________________________________________ 7
5. Quá trình hấp thu thuốc liên quan đến đường dùng thuốc ___________________ 7
5.1. Đường uống ___________________________________________________ 7
5.2. Đường ngậm dưới lưỡi và ngậm trong khoang miệng (buccal absorption) ___ 9
5.3. Đường trực tràng _______________________________________________ 9
5.4. Hấp thu qua da _________________________________________________ 9
5.5. Hấp thu qua đường hô hấp _______________________________________ 10
5.6. Đường tiêm bắp _______________________________________________ 10
5.7. Tiêm tĩnh mạch ________________________________________________ 11
BÀI 2: PHÂN BỐ THUỐC ____________________________________________ 12
1. Quá trình phân bố thuốc ____________________________________________ 12
2. Thể tích phân bố __________________________________________________ 13
3. Các yếu tố ảnh hưởng đến phân bố thuốc _______________________________ 14
3.1. Đặc điểm về thuốc _____________________________________________ 14
3.2. Đặc điểm sinh lý _______________________________________________ 15
3.3. Đặc điểm bệnh tật ______________________________________________ 16
3.4.Cạnh tranh vị trí liên kết _________________________________________ 16
3.5. Các trường hợp phân bố thuốc đặc biệt _____________________________ 17
4. Tích lũy thuốc ____________________________________________________ 18
BÀI 3: CHUYỂN HÓA THUỐC ________________________________________ 19
1. Quá trình chuyển hóa thuốc _________________________________________ 19
1.1. Khái niệm ____________________________________________________ 19
1.2. Đặc điểm của quá trình chuyển hóa thuốc ___________________________ 19
2. Các giai đoạn chính của quá trình chuyển hóa thuốc ______________________ 20
2.1. Giai đoạn I của chuyển hóa thuốc _________________________________ 21
2.2. Giai đoạn II của chuyển hóa thuốc _________________________________ 22
3. Hiện tượng cảm ứng, ức chế enzym cytochrome P450 ____________________ 24
3.1. Hiện tượng cảm ứng enzym cytochrome P450 _______________________ 24
3.2. Hiện tượng ức chế enzym cytochrome P450 _________________________ 24
4. Chuyển hóa qua gan lần đầu (First-pass effect) __________________________ 26
5. Chuyển hóa thuốc bởi vi khuẩn đường ruột _____________________________ 27
6. Các yếu tố ảnh hưởng đến chuyển hóa thuốc ____________________________ 27
6.1. Yếu tố sinh-bệnh lý ____________________________________________ 27
6.2. Yếu tố ngoại lai _______________________________________________ 28
BÀI 4: THẢI TRỪ THUỐC ___________________________________________ 30
1.Khái niệm thải trừ thuốc ____________________________________________ 30
2. Thải trừ thuốc qua thận _____________________________________________ 30
3. Thải trừ thuốc qua mật _____________________________________________ 32
4. Các yếu tố ảnh hưởng đến thải trừ thuốc _______________________________ 32
4.1. Yếu tố sinh-bệnh lý ____________________________________________ 32
4.2. Yếu tố ngoại lai _______________________________________________ 32
5. Độ thanh thải của thuốc và thời gian bán thải thuốc _______________________ 33
5.1. Độ thanh thải của thuốc (Clearance-Cl) _____________________________ 33
5.2. Thời gian bán thải của thuốc _____________________________________ 33
BÀI 5: CÁC MÔ HÌNH DƯỢC ĐỘNG HỌC CƠ BẢN _____________________ 35
1. Mô hình một ngăn với quá trình thải trừ bậc 1 ___________________________ 36
1.1. Truyền thuốc với tốc độ không đổi ________________________________ 36
1.2. Tiêm bolus một liều đơn _________________________________________ 38
1.3. Tiêm liều bolus lặp lại __________________________________________ 40
2. Các mô hình khác với mô hình một ngăn có quá trình đào thải bậc 1 _________ 43
2.1. Mô hình hai ngăn ______________________________________________ 43
2.2. Dược động học không tuyến tính (phụ thuộc liều) ____________________ 44
CHƯƠNG 2. SỰ THAY ĐỔI CÁC THÔNG SỐ DƯỢC ĐỘNG HỌC Ở BỆNH
NHÂN SUY GIẢM CHỨC NĂNG GAN, THẬN - ỨNG DỤNG ĐIỀU CHỈNH LIỀU
THUỐC TRÊN LÂM SÀNG _____________________________________________ 48
BÀI 6: SỰ THAY ĐỔI CÁC THÔNG SỐ DƯỢC ĐỘNG HỌC Ở BỆNH NHÂN
SUY GAN-ỨNG DỤNG ĐIỀU CHỈNH LIỀU THUỐC TRÊN LÂM SÀNG ___ 48
1. Bệnh gan và sự suy giảm chức năng gan _______________________________ 48
2. Vai trò của gan trong chuyển hóa và thải trừ thuốc _______________________ 48
3. Nồng độ thuốc trong huyết tương và tác dụng của thuốc ___________________ 52
4. Hiệu chỉnh liều lượng các thuốc chuyển hóa qua gan _____________________ 56
5. Một số nguyên tắc khi dùng thuốc cho người bệnh suy gan _________________ 57
BÀI 7: SỰ THAY ĐỔI CÁC THÔNG SỐ DƯỢC ĐỘNG HỌC Ở BỆNH NHÂN
SUY THẬN-ỨNG DỤNG ĐIỀU CHỈNH LIỀU THUỐC TRÊN LÂM SÀNG __ 59
1. Bệnh suy thận ____________________________________________________ 59
2. Vai trò của thận trong chuyển hóa và thải trừ thuốc _______________________ 60
3. Hướng dẫn hiệu chỉnh liều thuốc ở bệnh nhân suy thận ____________________ 63
3.1.Giữ nguyên khoảng cách đưa thuốc và giảm liều ______________________ 64
3.2. Giữ nguyên liều nhưng nới rộng khoảng cách đưa thuốc _______________ 64
3.3. Vừa giảm liều, vừa nới rộng khoảng cách đưa thuốc ___________________ 64
4. Một số nguyên tắc khi dùng thuốc ở bệnh nhân suy thận ___________________ 65
CHƯƠNG 3. GIÁM SÁT THUỐC ĐIỀU TRỊ (THERAPEUTIC DRUG
MONITORING -TDM) _________________________________________________ 67
BÀI 8: PHƯƠNG PHÁP GIÁM SÁT THUỐC ĐIỀU TRỊ __________________ 67
1. Các phương pháp giám sát thuốc điều trị _______________________________ 67
1.1. Phương pháp dược lực học _______________________________________ 67
1.2. Phương pháp dược động học _____________________________________ 68
2. Giám sát thuốc điều trị dựa vào nồng độ thuốc (Cp) ______________________ 69
2.1. Khái niệm ____________________________________________________ 69
2.2. Đối tượngcần giám sát thuốc điều trị _______________________________ 69
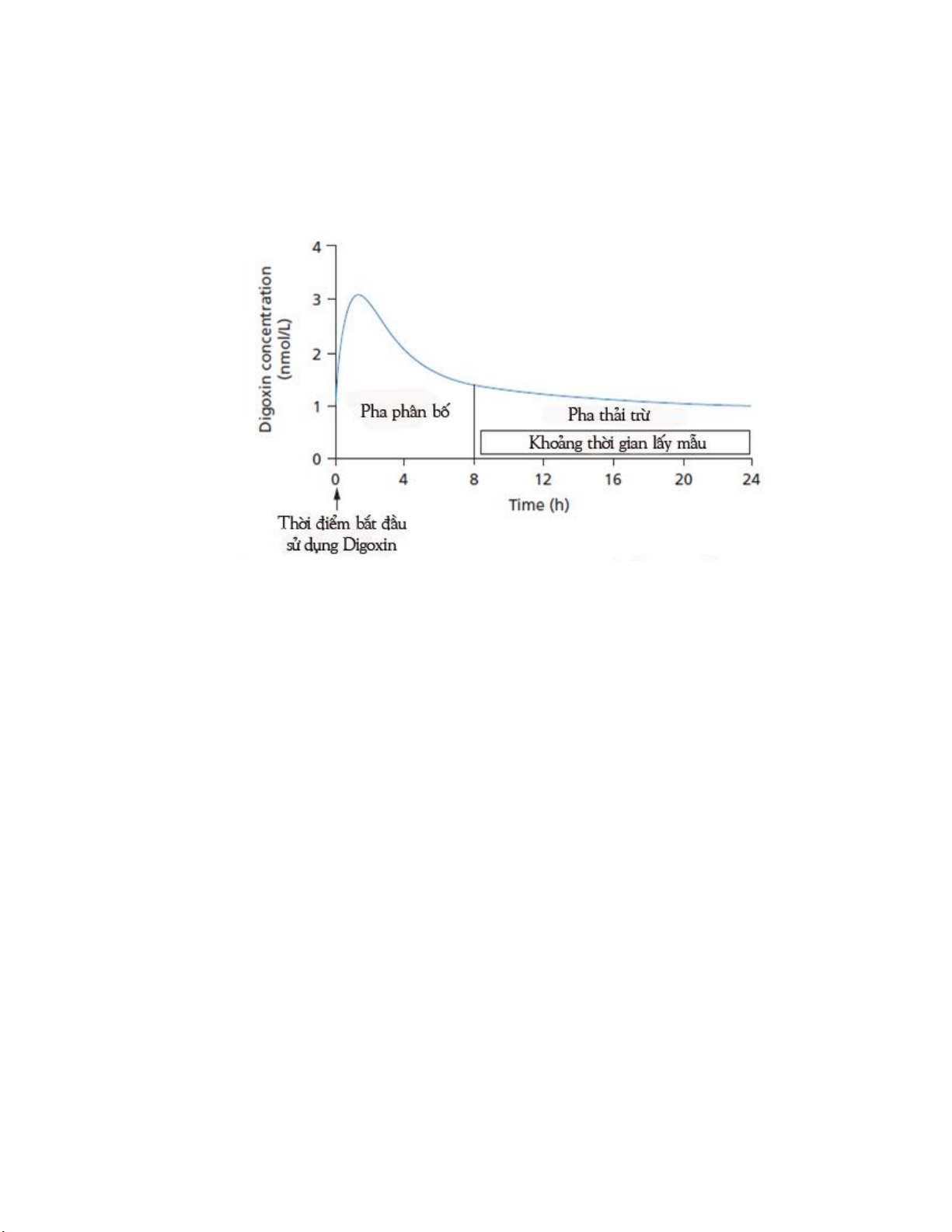
2.3. Những cân nhắc để diễn giải đúng nồng độ thuốc _____________________ 70
BÀI 9: GIÁM SÁT ĐIỀU TRỊ CỦA MỘT SỐ THUỐC ____________________ 80
1. Các thông tin cần thiết để theo dõi nồng độ thuốc Cp _____________________ 80
1.1. Về bệnh nhân _________________________________________________ 80
1.2. Về thuốc _____________________________________________________ 80
1.3. Về các thông số dược động học liên quan đến bệnh nhân _______________ 81
1.4. Các xét nghiệm ________________________________________________ 81
1.5. Thông tin về mẫu ______________________________________________ 82
2. Giám sát thuốc điều trị một số nhóm thuốc _____________________________ 83
2.1. Giám sát thuốc điều trị nhóm thuốc chống co giật _____________________ 83
2.2. Giám sát thuốc điều trị nhóm thuốc tác dụng trên tim mạch _____________ 84
2.3. Giám sát thuốc điều trị nhóm thuốc kháng sinh _______________________ 86
TÀI LIỆU THAM KHẢO _______________________________________________ 92
PHỤ LỤC ____________________________________________________________ 93
CHƯƠNG I. DƯỢC ĐỘNG HỌC
BÀI 1: HẤP THU THUỐC, SINH KHẢ DỤNG VÀ ĐƯỜNG DÙNG THUỐC MỤC TIÊU
1. Trình bày được các cơ chế vận chuyển thuốc chính trong cơ thể.
2. Trình bày được các khái niệm sinh khả dụng, tương đương sinh học, các công thức tính toán sinh khả dụng.
3. Phân tích được các yếu tố ảnh hưởng đến các quá trình hấp thu thuốc.
4. So sánh được ưu nhược điểm của các đường dùng thuốc khác nhau. NỘI DUNG
Để phát huy được tác dụng sinh học, thuốc trong cơ thể cần trải qua các biến đổi về
dược động học (Pharmacokinetic) và dược lực học (Pharmacodynamic). Dược động học
được định nghĩa là quá trình cơ thể tác dụng lên thuốc qua 4 giai đoạn Hấp thu
(Absorption), Phân bố (Distribution), Chuyển hóa (Metabolism) và Thải trừ
(Elimination). Dược lực học là quá trình thuốc thể hiện tác dụng dược lý lên cơ quan gây
ra đáp ứng lâm sàng (tác dụng điều trị và/hoặc độc tính) (Hình 1.1). Liều dùng Sinh khả dụng Dược Thuốc phân Nồng độ thuốc Thuốc chuyển động phối ở các mô trong tuần hoàn hóa và đào thải học Hệ số thanh thải
Phân phối Nồng độ thuốc ở nơi tác động Tác động dược lý Dược lực Đáp ứng lâm sàng học Độc tính Tác dụng điều trị Sử dụng
Hình 1.1. Các quá trình dược động lực học liên quan tới tác dụng của thuốc 1
1. Khái niệm hấp thu thuốc
Hấp thu thuốc là quá trình thuốc từ nơi đặt thuốc đi vào hệ tuần hoàn chung.
2. Cơ chế vận chuyển thuốc trong cơ thể
2.1. Vận chuyển thụ động (khuếch tán)
Thuốc từ nơi hấp thu muốn vào được hệ tuần hoàn cần có tính chất thân dầu để
vượt qua các hàng rào tế bào nằm ở niêm mạc đường tiêu hóa, cụ thể là vượt qua màng
sinh học phospholipid thân dầu hoặc nhờ các các protein màng từ nơi có nồng độ cao
(trong đường tiêu hóa) đến nơi có nồng độ thấp (hệ tuần hoàn). Vì thuốc di chuyển thuận
theo bậc thang nồng độ nên quá trình vận chuyển không cần năng lượng. Đa số các thuốc
hấp thu chủ yếu theo con đường này.
Như vậy, để thuốc vận chuyển qua con đường này, hai yếu tố ảnh hưởng quan trọng nhất là:
Khả năng thân dầu: thuốc càng thân dầu thì khả năng hấp thu càng tăng. Tuy nhiên,
các thuốc vẫn phải giữ một phần đặc tính thân nước để sau khi qua màng tế bào có
thể hòa tan để vượt qua nội bào thân nước.
Khả năng ion hóa: đa số các thuốc là các acid hay base yếu, vì vậy khả năng ion
hóa của thuốc phụ thuộc vào pH của môi trường nơi hấp thu.
Giả thuyết phân chia độ pH: Với acid yếu: Với base yếu:
Dựa vào công thức này cho thấy, đối với các thuốc acid yếu như aspirin ở trong
môi trường acid dịch vị thì nồng độ thuốc dạng không ion hóa HA (thân dầu) cao hơn nên
aspirin hấp thu tốt qua niêm mạc dạ dày và tá tràng. Còn đối với thuốc base yếu như
morphine thì ở trong môi trường kiềm ruột non thì nồng độ thuốc dạng không ion hóa
BOH (thân dầu) cao hơn nên thuốc hấp thu tốt ở ruột non. 2
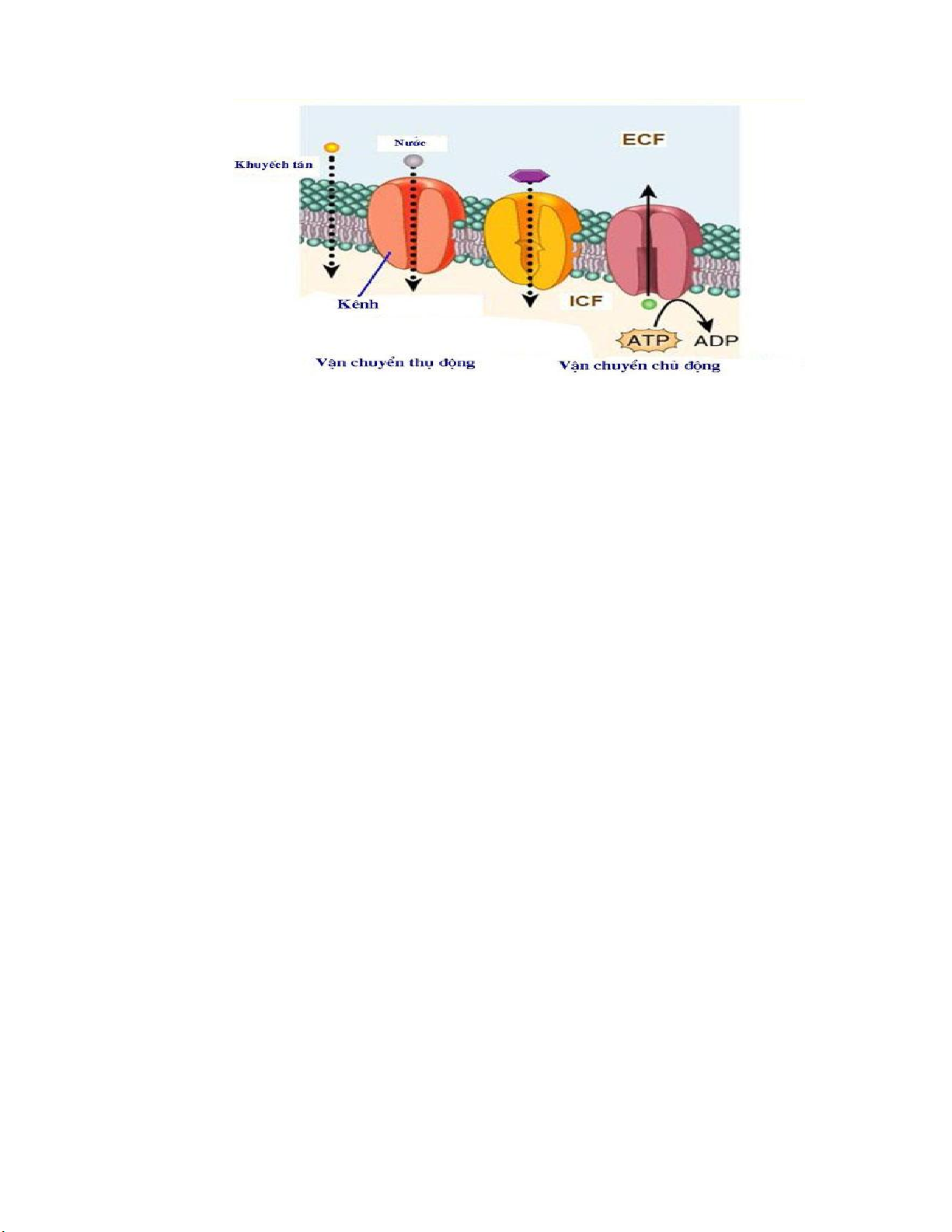
Hình 1.2. Các quá trình vận chuyển thuốc qua màng tế bào
Một số phân tử thân nước hoặc ion hóa với khối lượng phân tử nhỏ như nước, các
chất điện giải hay lithium có thể khuếch tán xuyên qua các kênh thân nước trong màng sinh học của tế bào.
2.2. Vận chuyển thụ động nhờ chất mang
Quá trình vận chuyển thụ động này không cần năng lượng nhưng cần sự có mặt
của chất mang nằm trong màng tế bào. Do đó, cơ chế này có các tính chất sau:
Tính bão hòa: quá trình vận chuyển này tăng đến một giới hạn bão hòa và không
thể tăng thêm được nữa.
Tính đặc hiệu: ứng với các thuốc khác nhau sẽ có những chất chuyên chở trung gian khác nhau.
Tính cạnh tranh: một số thuốc cùng vận chuyển bởi cùng một chất chuyên chở
trung gian sẽ cạnh tranh nhau.
2.2. Vận chuyển tích cực
Một số thuốc do phân tử lớn hoặc thân nước nên phải đi qua màng nhờ những chất
vận chuyển trung gian (carrier). Chất vận chuyển trung gian là các protein có sẵn trên
màng tế bào tạo nên những kênh xuyên màng tế bào tạo thuận lợi cho phân tử thuốc đi
qua. Quá trình vận chuyển tích cực này cần tiêu tốn năng lượng nên thuốc có thể được
vận chuyển ngược với bậc thang (gradient) nồng độ và tốc độ hấp thu không phụ thuộc
vào nồng độ thuốc. Vận chuyển tích cực có tính bão hòa, đặc hiệu và cạnh tranh. Ví dụ:
sự hấp thu levodopa sẽ giảm khi bị cạnh tranh bởi một số acid amine trong thức ăn. 3
3. Sinh khả dụng - Tương đương sinh học - Tiền thuốc - Thuốc gốc và Thuốc biệt dược 3.1. Sinh khả dụng
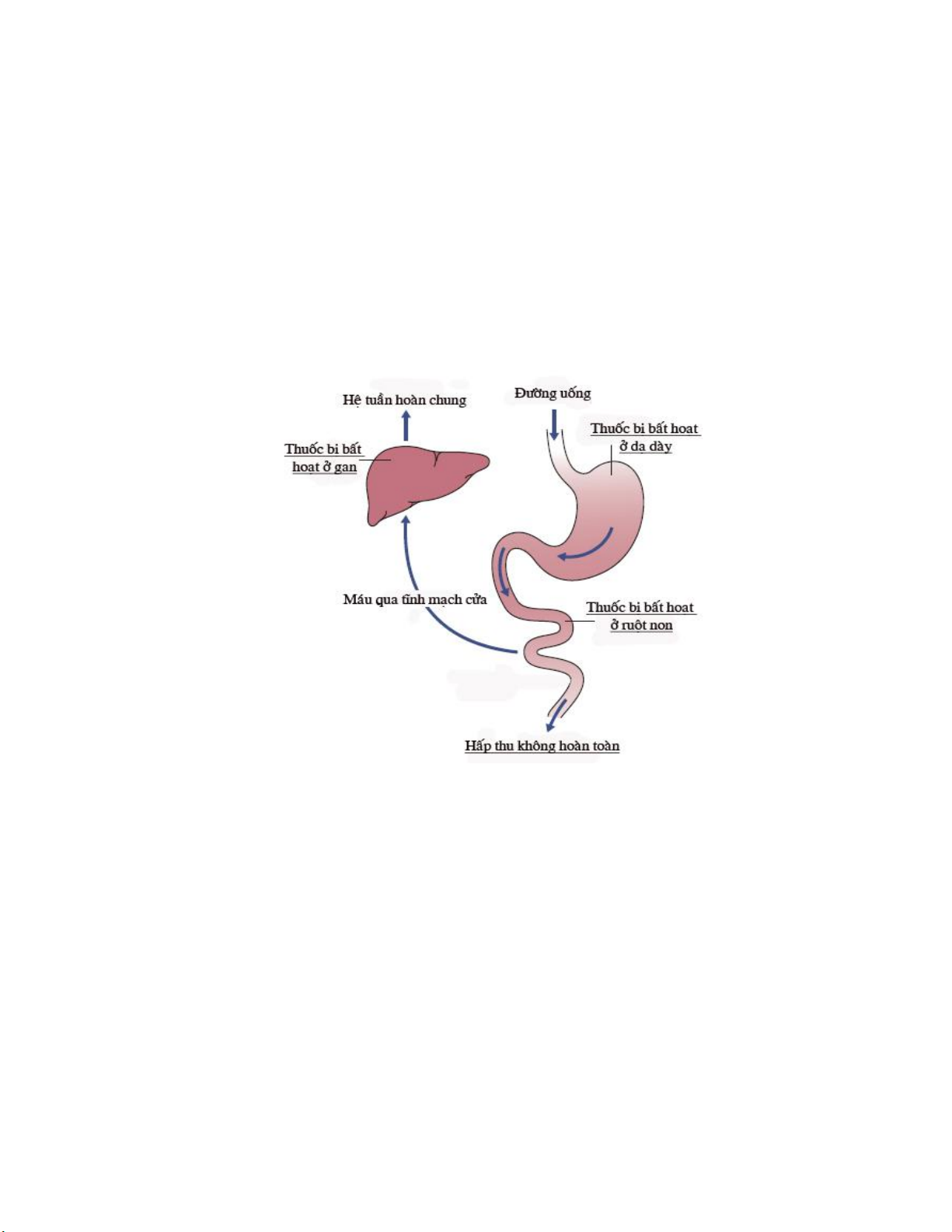
Thuốc phải đi vào hệ tuần hoàn để gây ra tác dụng dược lý toàn thân. Tuy nhiên,
trừ trường hợp tiêm truyền đường tĩnh mạch, hầu hết các thuốc đều hấp thu không hoàn
toàn vì những lý do sau: (1) thuốc bị bất hoạt trong ống tiêu hóa do acid dạ dày, enzyme
hay vi khuẩn ở đường tiêu hóa; (2) hấp thu không hoàn toàn; (3) quá trình chuyển hóa qua
gan lần đầu trong ống tiêu hóa và gan trước khi vào hệ tuần hoàn; (4) thuốc bị vận chuyển
ngược trở lại đường tiêu hóa (Hình 1.3).
Hình 1.3. Quá trình t
huốc bị bất hoạt trong cơ thể a. Định nghĩa
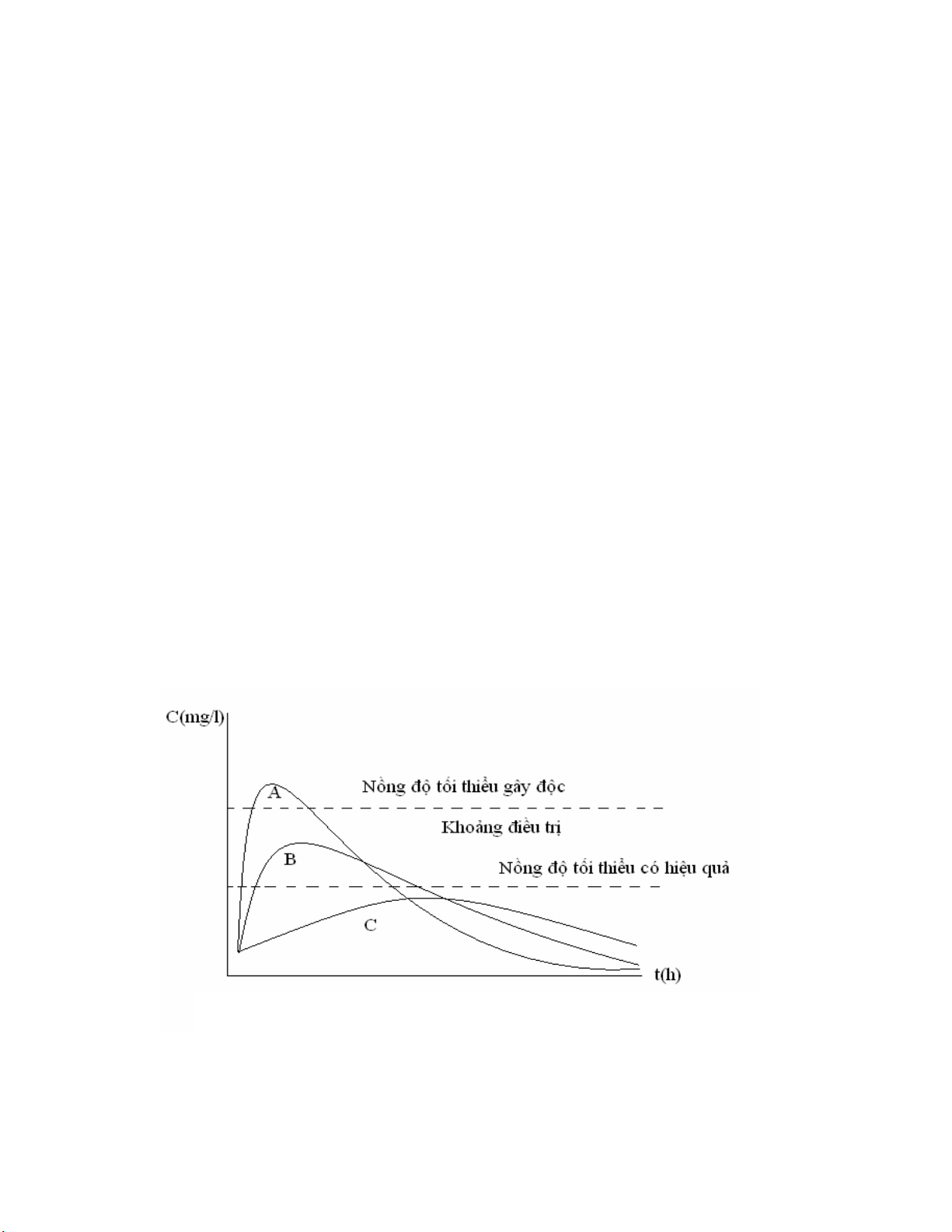
Sinh khả dụng (F) của một thuốc là phần thuốc vào được hệ tuần hoàn so với liều
thuốc được dùng ban đầu và tốc độ thuốc vào được hệ tuần hoàn (biểu hiện thông qua
Tmax là khoảng thời gian cần thiết để thuốc đạt nồng độ tối đa trong máu và Cmax là
nồng độ tối đa của thuốc trong máu).
b. Công thức tính sinh khả dụng
Sinh khả dụng tuyệt đối
Sinh khả dụng của một thuốc có thể được tính toán thực nghiệm bằng cách tính
diện tích dưới đường cong (AUC) của đồ thị biểu thị nồng độ thuốc theo thời gian trong 4
máu sau khi dùng liều Dpo qua một đường dùng thuốc nhất định (ví dụ như đường uống)
và liều Div thuốc đó khi dùng qua đường tĩnh mạch.
Nếu thuốc được đưa qua đường tĩnh mạch (IV) thì Ftđ=1.
Nếu thuốc được chỉ định ngoài đường tĩnh mạch thì luôn có một lượng thuốc nhất
định bị tổn hao khi đi từ vị trí hấp thu vào máu hoặc bị mất hoạt tính khi đi qua
gan, do đó Ftđ luôn <1.
Với những thuốc không thể dùng đường tĩnh mạch, người ta có thể dùng dạng
thuốc lỏng (dung dịch, hỗn dịch uống) để so sánh. Giá trị Ftđ:
< 50%: dạng thuốc thường khó đạt yêu cầu điều trị khi bệnh nặng
= 50%: là chấp nhận được trong điều trị.
> 80%: có thể coi khả năng thâm nhập của thuốc vào máu xấp xỉ đường tĩnh mạch.
Sinh khả dụng tương đối
Sinh khả dụng tương đối là tỷ lệ so sánh giữa 2 giá trị AUC của cùng 1 hoạt chất,
cùng 1 đường đưa thuốc, cùng 1 mức liều nhưng của 2 nhà sản xuất khác nhau hoặc của 2 dạng bào chế khác nhau.
Sinh khả dụng tương đối thường được sử dụng nhằm so sánh thuốc của một nhà
sản xuất nào đó với một thuốc đang lưu hành có uy tín trên thị trường (thường là dạng
uống) hoặc của một dạng viên với thuốc uống dạng lỏng.
3.2. Tương đương sinh học
Hai chế phẩm của cùng một hoạt chất, cùng liều dùng, cùng đường đưa thuốc được
coi là tương đương sinh học khi 3 đại lượng (F, Tmax, Cmax) này dao động ở mức độ cho
phép (80-125%). Hình 1.4 minh họa sự khác nhau về nồng độ thuốc trong huyết tương
theo thời gian của 3 dạng bào chế của cùng một dược chất. 5
3.3. Tiền thuốc - Thuốc gốc và thuốc biệt dược
Rất nhiều yếu tố trong quá trình sản xuất một chế phẩm thuốc ảnh hưởng đến sinh
khả dụng của chế phẩm thuốc đó. Ở Úc, trong hai năm 1968 và 1969, xảy ra nhiều trường
hợp ngộ độc phenytoin khi nhiều bệnh nhân dùng biệt dược phenytoin trong đó đã thay tá
dược calcium sulfate bằng lactose gây tăng sinh khả dụng thuốc và tăng độc tính. Sinh
khả dụng không chỉ khác nhau giữa các thuốc, các chế phẩm của cùng một thuốc mà còn
khác nhau giữa các cá thể mắc bệnh. Tuy nhiên sự khác nhau về sinh khả dụng không
nhất thiết luôn dẫn đến sự khác nhau quan trọng về tác dụng lâm sàng.
Một câu hỏi quan trọng trong thực hành lâm sàng là kê đơn thuốc bằng tên thuốc
gốc hay bằng tên biệt dược. Nếu bác sĩ kê đơn bằng tên biệt dược, dược sĩ có thể đề nghị
thay thế bằng một biệt dược sản xuất bởi công ty khác được chứng minh là tương đương
sinh học nhưng giá thành thấp hơn. Tuy nhiên, dù hai chế phẩm được chứng minh là
tương đương sinh học nhưng chưa chắc đã tương đương về điều trị. Nên cần thận trọng
khi thay thế với các thuốc có hệ số điều trị hẹp (tức liều tối thiểu có tác dụng và liều tối
thiểu gây độc gần nhau) hay các thuốc giải phóng kéo dài. Khó mà có câu trả lời thỏa
đáng cho câu hỏi liên quan đến có nên thay thế thuốc gốc- thuốc biệt dược hay không.
Một số thuốc có hoạt tính dược lý tốt nhưng lại khó hấp thu và/hoặc phân bố vào
các mô nên thường được bào chế dưới dạng tiền thuốc (prodrug) có khả năng hấp thu
và/hoặc phân bố vào các mô. Sau khi vào cơ thể, tiền thuốc sẽ được các enzyme trong cơ
thể chuyển hóa thành thuốc có hoạt tính.
Hình 1.4. Đồ thị nồng độ thuốc trong huyết tương của 3 dạng bào chế A, B,
C có cùng một dược chất 6
4. Các yếu tố ảnh hưởng đến hấp thu thuốc
4.1. Các yếu tố sinh - bệnh lý
Tuổi, hoạt động thể chất, tư thế khi uống thuốc, tình trạng có thai, tiêu chảy, táo
bón, suy tim…có thể làm thay đổi khả năng hấp thu thuốc.
4.2. Các yếu tố ngoại lai
a. Thức ăn, nước uống
Thức ăn có thể làm thay đổi tốc độ và/hoặc cường độ hấp thu của một số thuốc.
Một số thuốc được hấp thu tốt hơn nếu được uống cách xa bữa ăn ví dụ thuốc kháng lao
trong khi một số khác lại có sinh khả dụng tốt hơn khi uống trong bữa ăn ví dụ như
nitrofurantoin, griseofulvin. Đồ uống chứa nhiều calci như sữa có thể phản ứng với một
số thuốc tạo ra các phức chất không có khả năng hòa tan và không thể được hấp thu như
kháng sinh nhóm tetracyclin. b. Thuốc dùng kèm
Tương tác thuốc có thể xảy ra khi sử dụng đồng thời nhiều thuốc khác nhau. Ví dụ
thuốc kháng acid được dùng trong điều trị loét dạ dày-tá tràng có thể làm giảm hấp thu
của các thuốc khác khi uống cùng một lúc do các thuốc này ngăn cản sự tiếp xúc thuốc
với ống tiêu hóa thông qua lớp màng nhầy hoặc thông qua thay đổi pH dạ dày-ruột và khả
năng ion hóa của thuốc khác.
Một số thuốc khác có thể thúc đẩy nhanh sự tháo rỗng của dạ dày và nhu động của
đường tiêu hóa hoặc ngược lại làm chậm quá trình này dẫn đến thay đổi khả năng hấp thu
của thuốc. Ví dụ, thuốc làm chậm sự tháo rỗng của dạ dày như nhóm thuốc kháng
cholinergic có thể làm tăng hấp thu của thuốc tan kém nhờ kéo dài thời gian thuốc lưu lại
trong dạ dày như digoxin hoặc ngược lại, làm giảm khả năng hấp thu của một số thuốc do
kéo dài thời gian thuốc lưu lại trong dạ dày trong khi thuốc đó lại bị ezyme dạ dày phá hủy như levodopa.
5. Quá trình hấp thu thuốc liên quan đến đường dùng thuốc 5.1. Đường uống
Thuốc dùng đường uống có thể có tác dụng tại chỗ trong đường tiêu hóa (như
thuốc kháng acid điều trị bệnh loét dạ dày-tá tràng) hoặc tác dụng toàn thân.
Các yếu tố ảnh hưởng đến hấp thu gồm:
Phẫu thuật cắt bỏ một phần dạ dày làm giảm hấp thu một số thuốc.
Bệnh dạ dày-ruột như bệnh celiac đường ruột, bệnh xơ nang (cystic fibrosis).
Sự hiện diện của thức ăn. 7
Chuyển hóa thuốc bởi vi khuẩn đường ruột: khi dùng kháng sinh phổ rộng thường
làm giảm lượng vi khuẩn đường ruột có liên quan đến sự thủy phân estrogen liên
hợp ở dạ dày ruột, dẫn đến giảm quá trình tái hấp thu qua phòng tuần hoàn gan-
ruột làm giảm hiệu lực của thuốc tránh thai đường uống.
Chuyển hóa bởi emzyme: ví dụ cytochrome P450 3A ở trọng nội mạc dạ dày – ruột.
Vận chuyển ngược lại vào ống tiêu hóa bởi protein vận chuyển thuốc P- glycoprotein (P-gp), ABCB1).
Nhu động dạ dày – ruột có vai trò trộn đều dung dịch thuốc trong ống tiêu hóa.
Thời gian dạ dày rỗng càng ngắn, thuốc được đẩy xuống ruột non càng nhanh, làm
sớm gia tăng tốc độ hấp thu.
Thức ăn làm chậm hấp thu vì cản trở thuốc xuống ruột non.
Lượng máu đến hệ mạch máu mạc treo chậm (suy tim) làm chậm hấp thu.
Một số yếu tố liên quan đến khả năng hấp thu thuốc tại dạ dày và ruột được trình
bày trong bảng dưới đây.
Bảng 1.1. Các yếu tố liên quan đến hấp thu thuốc tại dạ dày và ruột Yếu tố Dạ dày Ruột pH 1-2 8 Diện tích 1m2 200m2 Lượng máu đến 0,15l/ph 1l/ph Độ thẩm thấu Yếu Mạnh
Thuốc tác dụng kéo dài
Một số thuốc với thời gian bán thải ngắn thường cần phải uống nhiều lần trong
ngày gây bất tiện cho bệnh nhân và bệnh nhân thường khó tuân thủ điều trị. Vì vậy, người
ta thường thay thế các thuốc này bằng các chế phẩm với hoạt tính tương tự nhưng được
thiết kế làm chậm sự hấp thu thuốc bằng cách giải phóng thuốc từ từ trong ruột non. Giảm
số lần dùng thuốc trong một ngày giúp tăng khả năng tuân thủ điều trị cho bệnh nhân,
đồng thời làm giảm tác dụng không mong muốn của thuốc do làm giảm nồng độ đỉnh
trong huyết tương như đối với carbamazepin. Sự hấp thu của các chế phẩm tác dụng kéo
dài thường không hoàn toàn, cho nên tương đương sinh học giữa các chế phẩm tác dụng
kéo dài là một yêu cầu bắt buộc. Các hạn chế khác của chế phẩm tác dụng kéo dài là: (1) 8
thời gian vận chuyển thuốc qua ruột non khoảng 6 tiếng, do đó liều hàng ngày của thuốc
có thể gây ra nồng độ đáy quá thấp; (2) lòng ruột quá hẹp hoặc nhu động ruột chậm như ở
người già hoặc một số thuốc dùng kèm (thuốc chống trầm cảm ba vòng, nhóm opiate) có
thể gây tích lũy thuốc tại nơi hấp thu, gây tổn thương niêm mạc đường tiêu hóa; (3) quá
liều với chế phẩm giải phóng kéo dài thường khó xử lý bởi vì sự hấp thu vẫn diễn ra sau
khi đã ngừng thuốc; (4) chế phẩm này không được bẻ, chia nhỏ; (5) giá thành cao.
5.2. Đường ngậm dưới lưỡi và ngậm trong khoang miệng (buccal absorption)
Thuốc được ngậm trong miệng để điều trị các rối loạn tại chỗ như hầu họng hoặc
niêm mạc miệng như viên ngậm hydrocortisone.
Đường ngậm dưới lưỡi có một lợi thế đặc biệt do thuốc hấp thu trực tiếp và nhanh
chóng vào hệ tuần hoàn qua hệ tĩnh mạch dưới lưỡi, không phải qua hệ niêm mạc đường
tiêu hóa nên không bị chuyển hóa qua gan lần đầu. Glyceryl trinitrate và fentanyl được
dùng dưới lưỡi vì những lý do này. Đường dùng này thường cho tác dụng ngắn và không
được nuốt viên thuốc.
Viên thuốc dùng ngậm trong khoang miệng cung cấp nồng độ trong huyết tương
ổn định hơn và thuốc được giữ trong khoang miệng giữa môi và lợi cho đến khi thuốc được tan hoàn toàn.
5.3. Đường trực tràng
Thuốc được đưa bằng đường trực tràng nhằm đạt tác dụng tại chỗ (ví dụ điều trị
viêm ruột thẳng) hoặc được chỉ định cho một số đối tượng đặc biệt khi không sử dụng
được các đường đưa thuốc khác (ví dụ paracetamol dành cho trẻ nhỏ). Lợi thế của đường
dùng này để có tác dụng toàn thân là: (1) tránh tiếp xúc với acid dạ dày và enzyme đường
tiêu hóa nên giảm tỷ lệ bị phá hủy; (2) tránh một phần chuyển hóa qua gan lần đầu; (3)
phù hợp với các đối tượng không sử dụng được các đường đưa thuốc khác. Diazepam
dùng đường trực tràng hữu ích trong trường hợp kiểm soát cơn động kinh ở trẻ em.
Metronidazole được hấp thu tốt qua đường trực tràng và rẻ hơn dùng đường tiêm tĩnh
mạch. Tuy nhiên, thuốc dùng đường trực tràng có thể gây kích ứng tại chỗ nghiêm trọng. 5.4. Hấp thu qua da
Một số thuốc được bôi tại chỗ để điều trị các bệnh ngoài da. Trong trường hợp này
hấp thu vào hệ tuần hoàn qua da gây tác dụng không mong muốn của thuốc, ví dụ dùng
glucocorticoid tại chỗ ở trẻ em. Dùng thuốc bôi qua da cũng có thể được dùng để gây tác
dụng toàn thân như miếng dán fentanyl để điều trị giảm đau. Da là một cấu trúc chống
thấm của cơ thể nên quá trình thuốc thấm qua da khác hoàn toàn với quá trình hấp thu qua
đường tiêu hóa. Yếu tố ảnh hưởng đến sự hấp thu của thuốc qua da gồm: (1) tình trạng
của da (sinh lý hay bệnh lý); (2) tuổi: trẻ em thường có tính thấm qua da cao hơn người 9
lớn; (3) vùng của da: Bàn chân < cẳng tay hóa của tế bào sừng: Tăng mức độ hydrate hóa tăng hấp thu nên một số trường hợp bịt kín
vùng bôi thuốc bằng các màng plastic để tăng hấp thu; (5) đặc tính lý hóa của thuốc: tính
thân lipid làm tăng hấp thu, giảm kích thước hạt làm tăng hấp thu và dung dịch hấp thu tốt
hơn dạng khác; (6) diện tích bề mặt dùng thuốc: Điều này cực kì quan trọng ở trẻ sơ sinh
vì trẻ sơ sinh thường có tỷ lệ diện tích bề mặt cơ thể/thể tích lớn. Thuốc dùng qua da để
có tác dụng toàn thân có lợi thế là tránh được chuyển hóa qua gan lần đầu. Tuy nhiên
miếng dán thường đắt hơn. Một số miếng dán dùng tác dụng toàn thân như miếng dán
nicotine cai nghiện thuốc lá, miếng dán scopolamine chống nôn khi đi tàu xe.
5.5. Hấp thu qua đường hô hấp
Phổi là nơi lý tưởng để hấp thu thuốc từ pha khí bởi vì diện tích bề mặt tiếp xúc
của phổi rất lớn gần 60m2. Thuốc điều trị hen như nhóm corticosteroid (ví dụ:
beclomethasone), nhóm cường giao cảm β2 adrenergic (ví dụ: salbutamol, salmeterol),
nhóm đối kháng phó giao cảm (ví dụ: ipratropium) được bào chế dạng khí dung để có tác
dụng tại chỗ trên khí quản. Kháng sinh dạng xịt cũng được dùng điều trị một số bệnh như
nang xơ hóa (cystic fibrosis) và nhiễm khuẩn Pseudomonas tái phát ở trẻ em. Đặc tính lý
hóa của thuốc giúp hạn chế hấp thu thuốc vào hệ tuần hoàn. Ví dụ ipratropium là dạng ion
thân nước, hấp thu kém vào hệ tuần hoàn nên giảm tác dụng không mong muốn so với
atropine dùng đường toàn thân. Một phần lớn liều dùng dạng hít của salbutamol không
vào phổi mà bị nuốt vào đường tiêu hóa và bị bất hoạt tại đó, gây tác dụng không mong
muốn toàn thân như run chi, tuy nhiên tác dụng này không đáng kể. Ngoài ra, các thuốc
gây mê và insulin cũng được bào chế dưới dạng khí dung.
5.6. Đường tiêm bắp
Nhiều thuốc được hấp thu tốt qua đường tiêm bắp. Tốc độ hấp thu phụ thuộc vào
khối lượng cơ. Có thể làm tăng sự phân tán thuốc bằng cách xoa bóp nơi tiêm trước khi
tiêm. Sự vận chuyển thuốc từ nơi tiêm vào hệ tuần hoàn phụ thuộc mật độ mạch máu
trong cơ và có sự khác nhau giữa các vị trí (cơ delta vai > cơ đùi > cơ mông). Dòng máu
tới cơ tăng nhờ tập thể dục nên tốc độ hấp thu tăng sau khi tập thể dục. Trái lại, sốc, suy
tim và các bệnh lý khác gây giảm dòng máu đến cơ làm giảm hấp thu thuốc.
Thuốc đòi hỏi phải thân nước nhất định để có thể hoà tan thành dung dịch tại nơi
tiêm cho đến khi hấp thu xảy ra. Do đó, một số thuốc tồn tại dạng tinh thể như phenytoin,
diazepam và digoxin khó hấp thu khi tiêm bắp nên cần tránh. Tuy nhiên, trong một số
trường hợp, người ta tận dụng sự hấp thu chậm khi muốn kéo dài tác dụng của thuốc. Ví
dụ: dạng este hóa của fluphenazine dùng để tiêm bắp, do đó thuốc cần được thủy phân để
giải phóng hoạt chất tự do có hoạt tính trước khi hấp thu, quá trình thủy phân làm chậm
hấp thu thuốc, kéo dài tác dụng của thuốc, tăng khả năng tuân thủ điều trị cho bệnh nhân. 10
Tuy nhiên, tiêm bắp có một số hạn chế sau: (1) đau: đặc biệt khi tiêm thể tích lớn,
vì vậy thể tích tiêm thường nên không quá 5ml; (2) gây áp xe vô khuẩn nơi tiêm ví dụ với
paraldehyde;(3) enzyme được giải phóng từ cơ làm tăng nồng độ creatinine
phosphokinase trong huyết tương có thể gây nhầm lẫn trong chẩn đoán; (4) tác dụng
không mong muốn nghiêm trọng của thuốc có thể bị kéo dài bởi vì không thể dừng hấp
thu thuốc khi đã lỡ tiêm thuốc; (5) với một số thuốc, tiêm bắp ít hiệu quả hơn dùng đường
uống; (6) hình thành ổ tụ huyết; (7) gây liệt dây thần kinh hông sau khi tiêm mông, có thể
tránh tác dụng phụ này bằng cách tiêm vào góc phần tư phía trên bên ngoài của mông.
5.7. Tiêm tĩnh mạch
Lợi thế khi tiêm tĩnh mạch gồm: (1) tác dụng nhanh ví dụ tiêm morphin để giảm
đau hay tiêm furosemid ở bệnh nhân phù nặng; (2) tránh sự chuyển hóa qua gan lần đầu ví
dụ truyền glyceryl trinitrat ở bệnh nhân đau thắt ngực không ổn định; (3) dùng cho thuốc
không hấp thu qua đường uống như nhóm aminoglycoside (gentamicin), heparin. Hoặc
các thuốc khi tiêm bắp quá đau hoặc quá độc; (4) truyền tĩnh mạch dễ kiểm soát, có thể
điều chỉnh tốc độ truyền đối với các thuốc có thời gian bán thải ngắn ví dụ: nitroprussid natri và epoprostenol.
Hạn chế của tiêm tĩnh mạch gồm: (1) một khi đã tiêm thuốc không thể thu hồi; (2)
nếu thuốc được tiêm quá nhanh gây tăng nồng độ thuốc đột ngột; (3) những thuốc gây độc
tế bào như thuốc trị ung thư (vincristine, doxorubicin) khi tiêm tĩnh mạch không được
phép rò rỉ khỏi tĩnh mạch vì có thể gây ra các tổn thương tại chỗ nghiêm trọng và gây đau;
(4) tiêm vô ý vào trong lòng động mạch có thể gây co thắt động mạch và gây hoại tử
ngoại vi; (5) gây tắc mạch do các phần tử lạ hay khí, nhiễm trùng máu hoặc huyết khối.
Ngoài ra còn có các đường dùng thuốc khác: mũi, mắt, tai, âm đạo, dịch não tủy…
Câu hỏi lượng giá
1. Sự khác nhau giữa cơ chế vận chuyển thụ động và tích cực là gì?
2. Trình bày sinh khả dụng của thuốc. Các công thức tính sinh khả dụng của thuốc?
3. Nêu các yếu tố ảnh hưởng đến hấp thu thuốc.
4. Trình bày một số đặc điểm hấp thu của một số đường dùng thuốc phổ biến. Ca lâm sàng
Một trẻ nhỏ 8 tháng tuổi chậm lớn, nhập viện để kiểm tra sức khỏe, bác sĩ chẩn
đoán có những biểu hiện của hội chứng Cushing. Bà mẹ cho biết thường tự ý mua
clobetasone bôi vào vùng đeo tã để điều trị nấm eczema cho trẻ. Kiểm tra không có dấu
hiệu tăng sản glucocorticoid nội sinh.
Câu hỏi: Vần đề liên quan đến sử dụng thuốc là gì? Cách thức xử lý? 11
BÀI 2: PHÂN BỐ THUỐC MỤC TIÊU
1. Trình bày được khái niệm, đặc điểm của quá trình phân bố thuốc.
2. Giải thích được ý nghĩa của thể tích phân bố.
3. Phân tích được các yếu tố ảnh hưởng đến phân bố thuốc
4. Trình bày quá trình phân bố thuốc vào hệ thần kinh trung ương và qua nhau thai. NỘI DUNG
1. Quá trình phân bố thuốc a. Khái niệm
Phân bố là quá trình thuốc phân bố vào trong các cơ quan cơ thể: mô mỡ, cơ,
xương, thần kinh trung ương…
b. Đặc điểm của quá trình phân bố thuốc
Thuốc sau khi được hấp thu vào hệ tuần hoàn, thuốc tồn tại ở hai dạng trong đó
dạng liên kết với protein huyết tương chủ yếu là với albumin, globulin và dạng tự do,
không liên kết với protein huyết tương. Chỉ có những phân tử thuốc ở dạng tự do mới
phân bố đến các cơ quan đích gây ra tác dụng dược lý. Hai dạng này tồn tại trạng thái cân
bằng động thuận nghịch nghĩa là khi dạng tự do trong máu giảm do thuốc ở dạng tự do
hấp thu từ máu dần vào các mô và cơ quan hoặc bị đào thải, thuốc dạng liên kết thuốc-
protein trong máu có xu hướng giải phóng khỏi liên kết với protein để chuyển thành dạng
tự do để bù đắp và ngược lại, chính vì vậy dạng thuốc liên kết với protein huyết tương
được coi như “kho dự trữ” của thuốc trong máu. Mức độ kết hợp tùy thuộc hằng số liên
kết K’a, nồng độ thuốc và nồng độ protein trong huyết tương, quá trình liên kết này có
tính bão hòa nghĩa là khả năng liên kết tăng đến một mức độ nhất định và không thể tăng thêm được nữa. K’a Thuốc + Protein Thuốc-Protein 12 2. Thể tích phân bố a. Khái niệm
Thể tích phân bố (Vd) là một thể tích lý thuyết, tính bằng đơn vị lít (l) để diễn tả
khả năng phân bố thuốc vào toàn bộ các mô, các cơ quan, đặc biệt ở những nơi mà thuốc
có thể gắn với các thụ thể (Receptor) để gây ra tác dụng dược lý.
Thể tích phân bố được định nghĩa là thể tích ảo để phân phối một liều thuốc D tiêm
trực tiếp vào tĩnh mạch để đạt được nồng độ thuốc trong huyết tương nào đó (Cp). Do đó: Vd = D/Cp
b. Ý nghĩa của thể tích phân bố
Trước hết, tìm hiểu ví dụ sau để hiểu vì sao Vd lại được gọi là thể tích ảo. Nếu
300mg một chất được hòa tan vào một ngăn hình hộp chứa một thể tích nước Vd và nồng
độ chất trong dung dịch được xác định là 0,1mg/ml thì thể tích phân bố Vd = 300/0,1 = 3 00ml.
Tuy nhiên, nếu một phần chất đó bị hấp thu mạnh vào thành của hình hộp (giả sử
90% chất đó bị hấp thu vào thành) làm nồng độ chất hòa tan trong dung dịch giảm còn
0,01mg/ml. Khi đó Vd = 300/0,01 = 30 000ml. Như vậy, phần thuốc càng hấp thu vào
thành càng lớn thì Vd càng tăng, trong khi thực tế ngăn hình hộp đó vẫn chỉ chứa 3 000ml nước mà thôi.
Có thể liên tưởng ví dụ trên với quá trình diễn ra trong cơ thể, nếu giả sử khi tiêm
nhanh 300mg thuốc vào tĩnh mạch, thuốc được hòa tan ngay vào huyết tương và thuốc
chưa kịp thải trừ. Nếu thuốc đó chỉ hòa tan trong huyết tương thì Vd của thuốc bằng với
Vd của huyết tương (xấp xỉ 3l ở người trưởng thành). Nhưng nếu thuốc được phân phối
từ huyết tương vào các cơ quan/mô, thì Vd sẽ tăng dần lên. Các thuốc phân phối vào các
cơ quan/mô càng mạnh thì Vd càng cao. Không có giới hạn tối đa của Vd.
Vdthuốc ~ VHT = 3l (ở bệnh nhân nặng trung bình 70kg): Thuốc chỉ ở trong ngăn
huyết tương vì có phân tử lượng lớn, khó vượt qua màng tế bào ví dụ heparin.
Vd thuốc ~ Vdịch ngoại bào = 16l (ở bệnh nhân nặng trung bình 70kg ): Thuốc chỉ
phân phối trong dịch ngoại bào vì ít tan/lipid; khó đi vào tế bào, đặc biệt là hệ thần
kinh trung ương, ví dụ tubocurarin.
Vd thuốc > Vddịch cơ thể = 46l (ở bệnh nhân nặng trung bình 70kg ): Thuốc dễ
tan trong lipid nên dễ qua màng tế bào gắn vào protein mô nhiều hơn protein huyết
tương. Các thuốc này không thể loại khỏi cơ thể bằng thẩm phân máu khi quá liều
ví dụ lidocain, digoxin, imipramin. 13
Bảng 2.1. Thể tích phân bố của một số thuốc Tên thuốc
Thể tích phân bố (Vd) (l) Haloperidol 20 Chlorpromazin 15 Digoxin 6 Propranolol 4 Diazepam 2 Tamicin 0,3 Phenylbutazol 0,1
Trong cơ thể, khả năng phân bố thuốc ảnh hưởng một phần đến thời gian có tác
dụng của thuốc, quyết định một phần đến số lần dùng thuốc trong ngày. Ví dụ sulfamide
chậm được gọi là “chậm” vì do thuốc gắn mạnh vào protein huyết tương, nên Vd lớn, dẫn
đến thời gian tác dụng kéo dài, làm giảm số lần dùng thuốc trong ngày.
3. Các yếu tố ảnh hưởng đến phân bố thuốc
3.1. Đặc điểm về thuốc
Khả năng thân dầu của thuốc giúp thuốc dễ vượt qua các màng tế bào để vào trong
các mô. Các thuốc gây mê có chỉ số lipid nước cao nên thấm được vào tế bào thần
kinh. Tác dụng gây mê càng nhanh khi chỉ số này càng cao. Do đó những người
béo phì cần một lượng thuốc gây mê lớn hơn và quá trình hồi phục sau gây mê
chậm hơn so với những người bình thường có cùng cân nặng.
Tính acid của thuốc (pKa): thuốc có tính base yếu, phần lớn không ở dạng ion hóa
dễ vượt qua hàng rào lipid vào sữa mẹ.
Ái lực của thuốc với protein huyết tương: khả năng gắn thuốc vào protein huyết
tương mạnh hay yếu là tùy loại thuốc:
Gắn mạnh (75-98%) như sulfamid chậm, rifampicin, lincomycin, quinin, phenylbutazon, phenytoin, diazepam, clopromazin, indomethacin,
dicoumarol, dogitoxin, furosemid, erythromycin, clopropamid…
Gắn yếu (1-8%) như barbital, sulfaguanidin, guanethidin…
Một số ít thuốc không gắn vào protein huyết tương như ure, glucose, uabain, lithium… 14
Trong điều trị, những liều đầu tiên của thuốc gắn mạnh vào protein huyết
tương thường phải đủ cao (liều tấn công) để bão hòa vị trí gắn, giúp cho liều
duy trì có thể đạt được tác dụng.
3.2. Đặc điểm sinh lý
Tuổi: khả năng liên kết với protein huyết tương thường kém ở trẻ em và người lớn
tuổi. Vì vậy cần thận trọng khi sử dụng thuốc có khả năng gắn kết mạnh với
protein như sulfamid, đặc biệt ở trẻ sơ sinh có biểu hiện tăng bilirubin trong máu.
Lưu lượng máu đến mô: phần thuốc tự do phân bố vào mô tức di chuyển từ ngăn
trung tâm (huyết tương) vào ngăn ngoại vi (mô) bằng cách xuyên qua các màng tế
bào của mô, theo cùng cơ chế như quá trình hấp thu thuốc. Quá trình này phụ
thuộc vào lưu lượng máu đến mô. Mô càng được tưới máu nhiều thì khả năng được
phân bố càng nhanh, thời gian để nồng độ thuốc trong mô đạt cân bằng với nồng
độ thuốc trong máu càng ngắn và ngược lại.
Bảng 2.2. Lưu lượng máu đến các mô Lưu lượng máu Mô % thể trọng % lưu lượng tim (ml/100g/ph) Thận 0,02 1 550 Thượng thận 0,4 24 450 Tuyến giáp 0,04 2 400 Gan 2 25 95 Tim 0,4 4 70 Não 2 15 55 Da 7 5 5 Cơ 40 15 3 Mô liên kết 7 1 1 Mỡ 15 2 1
Cấu tạo mô: một số mô có cấu tạo đặc biệt như một rào cản, ngăn thuốc xâm nhập
như hàng rào máu não, hàng rào nhau thai (xem phía dưới).
Người béo phì với sự phân bố mô mỡ, protein thay đổi nên có xu hướng thay đổi
khả năng phân bố của thuốc.
Phụ nữ mang thai với khối lượng nước tăng 50% nên liều của một số thuốc phải
tăng khi tính đến sự thay đổi này. 15
Trẻ sơ sinh: các thuốc có chỉ số lipid/nước thấp như theophylin, gentamicin khuếch
tán tốt vào các tổ chức có nhiều nước. Ở trẻ sơ sinh, nhất là trẻ đẻ thiếu tháng, tỷ lệ
nước của cơ thể chiếm tới 70-75%, do đó Vd của những thuốc này là rất lớn.
pH môi trường tại mô: thuốc phân bố đến các mô chịu ảnh hưởng sự khác biệt pH
giữa các mô đó. Ví dụ: tùy theo pH của mô mà thuốc ở dạng ion hay không ion,
nên thuốc vượt qua các màng phân cách giữa các mô dễ hay khó.
3.3. Đặc điểm bệnh tật
Suy tim ứ huyết làm giảm lưu lượng máu đến các mô làm giảm tốc độ và nồng độ
thuốc được phân bố, đồng thời suy tim sung huyết làm giảm lượng máu đến gan,
thận làm giảm lượng thuốc được thải trừ, gây tích lũy.
Các trường hợp bệnh lý ảnh hưởng đến lượng protein thông qua quá trình tổng hợp
protein, thay đổi cấu trúc protein, phân phối albumin trong và ngoài mạch, mức độ
thải trừ protein. Ví dụ, suy gan, hội chứng thận hư, bỏng, chấn thương, phẫu thuật,
suy dinh dưỡng…làm giảm lượng protein huyết tương gây tăng nồng độ thuốc tự
do trong máu, gây tăng tác dụng dược lý và/hoặc độc tính của thuốc, nên cần điều
chỉnh liều thuốc. Một số bệnh mãn tính làm thay đổi cấu trúc (chất lượng) protein
nên làm thay đổi K’a đối với thuốc.
Bảng 2.3. Các tình trạng bệnh làm giảm lượng protein huyết tương Tình trạng bệnh
Cơ chế làm giảm protein Bệnh gan Giảm tổng hợp
Chấn thương, phẫu thuật Tăng thoái hóa Bỏng
Phân phối albumin ra ngoài mạch Bệnh thận Đào thải nhiều protein
3.4.Cạnh tranh vị trí liên kết
Nếu dùng chung thuốc (A) với một loại thuốc (B) khác có liên kết với cùng một vị
trí trên protein: thuốc B cạnh tranh với thuốc A đẩy thuốc A ra khỏi vị trí liên kết, làm
nồng độ tự do của thuốc A tăng cao, gây độc tính. Đặc biệt khi thuốc A liên kết mạnh với protein (>95%).
Ví dụ 1: Trên bệnh nhân dùng thuốc chống đông kháng vitamin K đường uống là
thuốc có khả năng gắn kết mạnh với protein; khi bổ sung thuốc giảm đau kháng viêm
không steroid (NSAID) có thể gây ra sự cạnh tranh liên kết với protein huyết tương đẩy 16
thuốc kháng vitamin K ra khỏi huyết tương làm tăng nồng độ thuốc kháng đông tự do
trong máu, gây tác dụng không mong muốn xuất huyết.
Ví dụ 2: Sự cạnh tranh liên kết với protein còn xảy ra giữa thuốc với các chất nội
sinh trong cơ thể như bilirubin trong trường hợp bilirubin tăng bất thường trong máu.
Ví dụ 3: Bệnh nhân dùng tolbutamid để điều trị đái tháo đường, nay có đau khớp
dùng thêm phenylbutazon, phenylbutazon sẽ đẩy tolbutamid ra dạng tự do, gây hạ đường
huyết đột ngột. Vì vậy, trong điều trị khi phối hợp nhiều thuốc cần lưu ý vấn đề này.
3.5. Các trường hợp phân bố thuốc đặc biệt
a. Vận chuyển thuốc vào thần kinh trung ương
Tại đây, thuốc phải vượt qua 3 “hàng rào”:
Từ mao mạch não vào mô thần kinh (hàng rào máu-não)
Từ đám rối màng mạch vào dịch não tủy (hàng rào máu-màng não hay máu-dịch não tủy).
Từ dịch não tũy vào mô thần kinh (hàng rào dịch não tủy-não).
Như vậy, ở thần kinh trung ương, thuốc gặp những chướng ngại vật là thể liên kết
ở các khoảng gian bào và chân những tế bào sao, nên thuốc phải mất nhiều giờ, có khi
nhiều ngày mới đạt được cân bằng nồng độ máu/não, khác với cân bằng máu/cơ chỉ cần vài phút hoặc vài giây.
Sự vận chuyển thuốc qua hàng rào thần kinh trung ương còn phụ thuộc vào lứa
tuổi, trạng thái bệnh lý, đặc điểm của thuốc:
Ở trẻ nhỏ và trẻ sơ sinh lượng myelin còn ít, cấu trúc “hàng rào” chưa hoàn thiện
nên thuốc dễ khuếch tán vào não.
Penicillin không thấm qua được màng não bình thường, nhưng khi màng não bị
viêm, penicillin và nhiều thuốc khác có thể qua được.
Các thuốc có tính thân lipid, không bị ion hóa và không liên kết mới có khả năng
thấm qua, nhưng không ở lại lâu. Thuốc được vận chuyển qua hàng rào máu não
bằng vận chuyển thụ động hay tích cực hoặc nhờ chất vận chuyển. Các thuốc dễ
thấm qua hàng rào máu não: Kháng sinh betalactam, metronidazol, thuốc chống
trầm cảm, nhóm thuốc statin…
Thuốc thân nước, ion hóa nhiều khó vượt qua hàng rào máu-não. 17
b. Vận chuyển thuốc qua nhau thai:
Cấu tạo của nhau thai phức tạp, là hàng rào tự nhiên ngăn cản các chất từ tuần
hoàn mẹ sang thai nhi. Nhau thai còn chứa nhiều enzyme như cholinesterase, mono amin
oxidase, hydroxylase…có thể chuyển hóa một số thuốc, làm thuốc giảm tác dụng để bảo
vệ thai nhi. Tính thấm của màng mao mạch thai nhi tăng theo tuổi thai và sự thấm thuốc
cũng theo quy luật chung: thông thường, hàng rào nhau thai cho qua những thuốc thân
lipid như thuốc mê thiopental (cơ chế vận chuyển thụ động), thuốc có phân tử lượng nhỏ
như acid amin, glucose, các ion Ca++, Mg++, vitamin… (cơ chế vận chuyển tích cực),
thuốc ở dạng tự do. Huyết tương của thai nhi có tính acid hơn huyết tương cơ thể mẹ nên
các thuốc có tính base yếu dễ thấm qua như erythromycin. Trừ các thuốc tan trong nước
có trọng lượng phân tử lớn như dextran và các amin bậc 4 như galamin, neostigmin là
thuốc không qua được nhau thai, còn rất nhiều thuốc có thể vào được máu thai nhi, gây
độc tính cho thai nhi như thuốc nhóm điều trị ung thư, isotretinoin, phenobarbital,
sulfamid, morphin, fluoquinolon. 4. Tích lũy thuốc
Thuốc sau khi phân bố vào mô sẽ gắn với receptor của nó để tạo nên tác dụng dược
lý. Thuốc còn có thể tích lũy ở một bộ phận nào đó trong cơ thể hoặc được chuyển hóa
bởi enzyme chuyển hóa thuốc.
Thuốc tích lũy có thể bằng cách tạo liên kết cộng hóa trị với một số mô trong cơ
thể và được giữ lại hàng tháng đến hàng chục năm sau dùng thuốc (hoặc chất độc) có khi
chỉ sau một lần dùng, ví dụ thuốc DDT gắn vào mô mỡ, tetracyclin gắn vào những mô
đang calci hóa (sụn tiếp hợp, răng trẻ em), asen gắn vào tế bào sừng, lông, tóc…Thuốc
cũng có thể được vận chuyển tích cực vào mô để tích lũy ví dụ nồng độ quinacrin trong tế
bào gan khi dùng thuốc dài ngày có thể cao hơn nồng độ trong huyết tương vài trăm lần.
Câu hỏi lượng giá:
1. Trình bày khái niệm, đặc điểm của quá trình phân bố thuốc.
2. Giải thích ý nghĩa của thể tích phân bố.
3. Phân tích các yếu tố ảnh hưởng đến phân bố thuốc.
4. Trình bày quá trình phân bố thuốc vào hệ thần kinh trung ương và qua nhau thai.
5. Khi tiêm tĩnh mạch nhanh 0,5mg digoxin vào cơ thể một bệnh nhân nặng 70kg, đo
được nồng độ thuốc trong huyết tương là 0,78ng/ml. Tính Vd. 18
BÀI 3: CHUYỂN HÓA THUỐC MỤC TIÊU
1. Trình bày được khái niệm và đặc điểm của quá trình chuyển hóa thuốc.
2. Nêu được các giai đoạn chính của chuyến hóa thuốc.
3. Phân tích được ảnh hưởng của hiện tượng cảm ứng, ức chế enzyme cytochrome P450
lên quá trình chuyển hóa của thuốc.
4. Trình bày được quá trình chuyển hóa thuốc qua gan lần đầu và chuyển hóa thuốc do vi khuẩn ruột.
5. Phân tích được các yếu tố ảnh hưởng đến chuyển hóa thuốc. NỘI DUNG
1. Quá trình chuyển hóa thuốc 1.1. Khái niệm
Một thuốc được đào thải dễ dàng hơn qua các cơ quan (chủ yếu là thận) nếu thuốc
thân nước. Quá trình chuyển hóa thuốc là quá trình thuốc biến đổi về tính chất lý hóa học
trong cơ thể khi chịu tác động chủ yếu của các enzym chuyển hóa thuốc, thường tạo các
sản phẩm thân nước hơn giúp đào thải thuốc ra khỏi cơ thể dễ dàng hơn.
1.2. Đặc điểm của quá trình chuyển hóa thuốc
Sản phẩm sinh ra do quá trình chuyển hóa thuốc gọi là chất chuyển hóa của thuốc.
Chất chuyển hóa của thuốc thường không có tác dụng dược lý gọi là chất chuyển hóa bất
hoạt tính. Do đó, chuyển hóa thuốc là quá trình chính giúp làm mất hoạt tính của thuốc.
Ví dụ như quá trình oxy hóa phenytoin và rượu ethanol. Tuy nhiên, một số thuốc gọi là
tiền thuốc (prodrug), quá trình chuyển hóa tạo ra chất chuyển hóa có hoạt tính (ví dụ như
oxazepam, chất chuyển hóa còn hoạt tính của diazepam); thậm chí chất chuyển hóa có
hoạt tính mạnh hơn thuốc gốc (ví dụ như canrenone, chất chuyển hóa còn hoạt tính của
spironolactone). Trong một số trường hợp, quá trình chuyển hóa có thể tạo nên các chất
chuyển hóa gây độc (ví dụ như acetylisoniazide, chất chuyển hóa gây độc cho gan của
isoniazide). Một số quá trình chuyển hóa thuốc sản sinh chất chuyển hóa có hoạt tính với
thời gian bán thải dài hơn cả thuốc gốc, nên chất chuyển hóa có hoạt tính tích lũy chậm
hơn để đạt được trạng thái có nồng độ ổn định trong máu, nên phát huy tác dụng điều trị
muộn (ví dụ diazepam có thời gian bán thải là 20-50 giờ, trong khi chất chuyển hóa có
hoạt tính của nó là desmethyldiazepam có thời gian bán thải xấp xỉ 100 giờ).
Lưu lượng máu tới gan lớn và giàu các enzyme chuyển hóa thuốc nên gan đóng vai
trò quan trọng nhất trong việc chuyển hóa thuốc. Ngoài ra, một số thuốc còn được chuyển
hóa qua các cơ quan khác như ống tiêu hóa, phổi, thận... 19
2. Các giai đoạn chính của quá trình chuyển hóa thuốc
Hầu hết các thuốc tan trong lipid được chuyển hóa ít nhiều ở gan. Để thuận tiện, có
thể chia quá trình chuyển hóa thành 2 giai đoạn, thường xảy ra kế tiếp nhau. Giai đoạn I
như các phản ứng oxy hóa, phản ứng thủy phân và phản ứng khử thường qua trung gian là
hệ thống enzyme cytocrom P-450 (CYP ) được liên kết với màng của mạng lưới nội chất
bên trong tế bào gan. Phản ứng giai đoạn I thường làm bất hoạt thuốc hay chuyển hóa
thành sản phẩm có hoạt tính. Các phản ứng Giai đoạn II bao gồm các phản ứng liên hợp
glucuronide, acetatehoặc sulfate, cũng có thể được xúc tác qua trung gian là các enzyme
của tế bào gan. Các phản ứng chuyển hóa thuốc qua Giai đoạn II thường tạo thành các
chất chuyển hóa thân nước hơn nên dễ được thải trừ qua thận hoặc các phân tử thuốc
được chủ động thải trừ qua mật thông qua các protein vận chuyển như P- glycoprotein. Ví
dụ, phenytoin thường bị oxy hóa thành 4-hydrophenytoin, sau đó liên hợp với glucuronide
thành 4-hydroxyphenytoin-glucoronide và chất này được đào thải ra ngoài qua thận. Thuốc trong cơ thể
Chuyển hoá giai đoạn I (Phase I)
Chuyển hoá giai đoạn II (Phase II) Phản ứng oxy hoá Phản ứng acetyl hoá Phản ứng khử Phản ứng methyl hoá Phản ứng thuỷ phân Phản ứng sulphat hoá
Phản ứng liên hợp với acid glucuronic
Phản ứng liên hợp với glutathione
Phản ứng liên hợp với acid mercapturic
Thải trừ qua thận hoặc mật (a) 20
Hình 3.1. (a) Giai đoạn I và II của chuyển hóa thuốc; (b) Giai đoạn I và II của
chuyển hóa phenobarbital
2.1. Giai đoạn I của chuyển hóa thuốc
Gan là nơi quan trọng nhất để chuyển hóa thuốc. Màng lưới nội chất bên trong tế
bào gan đóng vai trò quan trọng nhất, ngoài ra ti thể và dịch bào tương cũng tham gia vào chuyển hóa thuốc.
a. Màng lưới nội chất
Màng lưới nội chất trơn trong tế bào gan chứa các đại gia đình enzyme cytochrome
P450 (CYP450), có hơn 50 CYP khác nhau ở người giúp chuyển hóa các chất ngoại lai
như thuốc, thuốc trừ sâu, các chất hóa học khác. Các phản ứng gồm phản ứng oxy hóa, khử và thủy phân. Phản ứng oxy hóa
Phản ứng oxy hóa gây thơm hóa, tách nhóm amin, tách nhóm alkylhoặc oxy hóa
lưu huỳnh. Những phản ứng này đều cần sự tham gia của NADP khử (nicotinamide
adenine dinucleotide phosphate), phân tử oxy và một hoặc vài nhóm protein CYP450
đóng vai trò oxy hóa cuối cùng trong chuỗi phản ứng oxy hóa.
CYP450 còn liên quan đến quá trình sinh tổng hợp các chất trung gian hoặc các
chất điều hòa quan trọng trong cơ thể. Ví dụ, enzyme tổng hợp hỗ trợ trong quá trình oxy
hóa acid arachidonic thành prostaglandin và thromboxane là CYP450. Phản ứng khử
Phản ứng khử đòi hỏi enzyme khử NADP-cytpchrome-c reductase hoặc enzyme
khử NAD-cytochrome b5 reductase.
Phản ứng thủy phân
Pethidine bị thủy phân thành acid meperidine bởi hoạt tính của men thủy phân
esterase ở màng tế bào gan.
b. Chuyển hóa thuốc không phải ở màng lưới nội chất 21 Phản ứng oxy hóa
Phản ứng oxy hóa ethanol thành acetaldehyde và chloral thành trichlorethanol
được xúc tác bởi enzyme ở dịch bào tương, là enzyme chuyển hóa vitamin A. Enzyme
monoamine oxidase (MAO) là một enzyme trong ti thể gắn ở màng ti thể giúp oxy hóa
đồng thời làm tách nhóm amin tạo thành aldehyde, acid carboxylic hoặc ketone. MAO
nằm trong tế bào gan, thận, ruột non, mô thần kinh và nó giúp chuyển hóa các
catecholamine (dopamine, noradrenaline, adrenaline), tyramine, phenylepherine, dẫn chất
tryptophan (5-hydrotryptamine và tryptamine). Oxy hóa các purine (như 6-mercatopurin
bị bất hoạt thành 6-thiouric acid) được thực hiện bởi enzyme xanthine oxidase. Phản ứng khử
Phản ứng khử bởi enzyme tạo ra các nối đôi như đối với thuốc methadone, naloxone.
Phản ứng thủy phân
Phản ứng thủy phân các ester nhờ xúc tác là các enzyme esterase xảy ra với nhiều
thuốc. Ví dụ như thủy phân acetylsalicylic (aspirin) thành salicylate, thủy phân enalapril thành enalaprilate.
2.2. Giai đoạn II của chuyển hóa thuốc
Phản ứng gắn acid amine
Glycine và glutamin là những acid amin chủ yếu liên quan đến phản ứng liên hợp
ở người. Glycine liên hợp với acid nicotinic và salicylate trong khi glutamine liên hợp với
p-aminosalicylate. Tổn thương tế bào gan làm giảm dự trữ các acid amin này, vì vậy phản
ứng liên hợp này bị hạn chế. Phản ứng gắn acid amin bị giảm.
Phản ứng acetyl hóa
Phản ứng liên hợp với nhóm acetyl nhờ acetyl coenzyme A xảy ra với một số
thuốc như isoniazid, hydralazine, procainamide. Phản ứng này xảy ra ở dịch bào tương và
ở bạch cầu, nội mạc dạ dày-ruột và gan.
Phản ứng liên hợp glucuronide
Phản ứng liên hợp giữa acid glucuronic và nhóm carboxyl liên quan đến quá trình
chuyển hóa bilirubin, salicylat và lorazepam. Một số bệnh nhân bị bệnh di truyền và thiếu
hụt khả năng phản ứng liên hợp glucuronid dẫn đến triệu chứng vàng da không tan huyết
trên lâm sàng do nồng độ bilirubin không liên hợp tăng cao (gọi là hội chứng Crigler-
Najjar). Những thuốc chuyển hóa qua con đường này làm trầm trọng thêm triệu chứng
vàng da trên bệnh nhân. Ngoài ra, phản ứng liên hợp O-glucuronide xảy ra với một số
thuốc như paracetamol và morphin. 22
Phản ứng methyl hóa
Phản ứng methyl hóa thường sử dụng S-adenosyl methionine như chất cung cấp
nhóm methyl cho thuốc có chứa nhóm thiol, hydroxyl, amino tự do. Enzyme catechol O-
methyltransferase là một enzyme của phản ứng methyl hóa có vai trò quan trọng trong
chuyển hóa sinh lý của cơ thể cũng như chuyển hóa thuốc. Enzyme này nằm trong dịch
bào tương, xúc tác vận chuyển nhóm methyl gắn vào các catecholamin, làm bất hoạt
noradrenamin, dopamin và adrenalin. Enzyme phenylethanolamin N-methyltransferase là
một enzyme khác đóng vai trò quan trọng trong chuyển hóa catecholamin. Enzyme này
gắn nhóm methyl vào gốc –NH2 của noradrenamin để tạo thành adrenalin ở tủy tuyến
thượng thận và tác dụng lên các amin nội sinh như phenylethanolamin và phenylephrin.
Enzym này được cảm ứng bởi corticosteroid. Dòng máu từ vỏ thượng thận đi vào tủy
thượng thận có nồng độ corticosteroid rất cao. Điều này giải thích vì sao hoạt tính của
enzym này trong tủy thượng thận rất cao.
Phản ứng sulphate hóa
Enzyme vận chuyển nhóm sulfate (sulphotransferase) nằm trong dịch bào tương
xúc tác phản ứng chuyển nhóm sulphuryl từ 3’-phosphoadenosine 5’-phosphosulphate
(PAPS) tới nhóm hydroxyl (-OH) và nhóm amin của thuốc. Trong điều kiện sinh lý,
sulphotransferase xúc tác phản ứng tạo heparin sulphate và chondroitin sulphate. Ngoài
ra, một số enzyme sulphotransferase khác nhau ở tế bào gan giúp chuyển hóa các chất khác.
Phản ứng liên hợp với acid mercapturic
Phản ứng liên hợp với acid mercapturic là phản ứng với cysteine trong nhóm 3
peptide Cys-Glu-Gly có trong phân tử glutathion. Phản ứng này rất quan trọng khi quá
liều paracetamol (acetaminophen), khi quá trình liên hợp thông thường với sulphate,
glucuronid của paracetamol đã bị quá tải, tạo ra chất chuyển hóa có độc tính cao là N-
acetyl-p-benzoquinone imine (NAPQI). NAPQI thông thường được khử độc bằng cách
liên hợp với glutathion khử. Vì vậy, những bệnh nhân bị ngộ độc paracetamol được chỉ
định N-acetyl cysteine hoặc methionin để tăng tổng hợp glutathion nội sinh.
Phản ứng liên hợp với glutathion
Naphthalen và một số sulfonamid phản ứng liên hợp với glutathion. Một chức
năng nội sinh của phản ứng liên hợp glutathion là hình thành một sulfidopeptide
leukotriene gọi là leukotriene (LT) C4. Nó được hình thành bằng cách liên hợp glutathion
và LTA4. LTA4 là một epoxid được tổng hợp từ acid arachidonic. LTC4 là một trong số
những leukotrien đóng vai trò quan trọng như một chất điều hòa co thắt khí quản trong
phản ứng sốc phản vệ và trong hen. 23
3. Hiện tượng cảm ứng, ức chế enzym cytochrome P450
Các CYP 450 có mặt ở gan và ruột. Các enzyme CYP 450 được phân chia thành
nhiều họ enzym (family) và phân họ enzym (subfamily) gồm các isoenzyme khác nhau,
có khoảng hơn 50 enzyme khác nhau. Các phân họ CYP chuyển hóa các chất khác nhau.
Tỷ lệ phần trăm các thuốc dùng trên lâm sàng chuyển hóa bởi mỗi isoenzyme CYP là:
CYP3A4 50%; CYP2D6 20%, CYP2C 20%; CYP1A2 2%; các CYP khác 6%.
3.1. Hiện tượng cảm ứng enzym cytochrome P450
Cảm ứng enzym là hiện tượng hoạt tính của enzym tăng lên, thường bởi tăng tổng
hợp enzym (hoặc ít phổ biến hơn bởi giảm thoái hóa enzym). Kết quả của quá trình cảm
ứng enzym là, khi hai hoặc nhiều thuốc được dùng đồng thời, nếu một thuốc là tác nhân
gây cảm ứng enzym còn thuốc kia chuyển hóa qua enzym đó, thì quá trình cảm ứng
enzym sẽ làm tăng quá trình chuyển hóa của thuốc khác và có thể dẫn đến giảm nồng độ
thuốc trong máu, gây thất bại điều trị.
Ví dụ: Rifampicin để điều trị bệnh lao có thể làm cho thuốc tránh thai đường uống
không còn hiệu quả do rifampicin làm cảm ứng enzym làm tăng chuyển hóa thuốc tránh thai.
Quá trình cảm ứng thường chậm, thông thường phải mất 1-2 tuần. Quá trình cảm
ứng enzym gây bởi một tác nhân nhất định là khác nhau rõ rệt giữa các cá thể, một trong
số nguyên nhân là do khác nhau về kiểu gen.
Các tác nhân gây cảm ứng enzym có thể là thuốc như nhóm thuốc chống động kinh
(phenobarbital, phenytoin, fosphenytoin, carbamazepin, primidon); thuốc kháng lao
(rifampicin, rifabutine, isoniazid); một vài thuốc kháng virus (efarirenz, nevirapine);
glucocorticosteroid (prednisone, dexamethasone); vài thuốc trị đái tháo đường (insulin,
pioglitazone). Tác nhân gây cảm ứng enzyme cũng có thể không phải là thuốc như thuốc
trừ sâu halogen hóa (đặc biệt là dichloro-diphenyl-trichloroethane (DDT) và gamma-
benzen hexachloride), thuốc diệt cỏ, các hydrocarbon vòng thơm nhiều vòng, các chất bảo
quản thức ăn, nicotine, ethanol và hyperforin có trong cây St John’s wort để điều trị trầm cảm.
3.2. Hiện tượng ức chế enzym cytochrome P450
Ức chế enzyme là một hiện tượng hoạt tính của enzym giảm đi do bị ức chế trực
tiếp bởi tác nhân gây ức chế. Kết quả của quá trình ức chế enzym là, khi hai hoặc nhiều
thuốc được dùng đồng thời, nếu một thuốc là tác nhân gây ức chế enzym còn thuốc kia
chuyển hóa qua enzym đó, thì quá trình ức chế enzym sẽ làm giảm quá trình chuyển hóa
của thuốc khác và có thể dẫn đến tăng nồng độ thuốc trong máu, gây tăng độc tính.
Tính đặc hiệu của quá trình ức chế enzym có khi là không hoàn toàn. Ví dụ,
warfarin và phenytoin cùng cạnh tranh chuyển hóa lẫn nhau, nên dùng hai thuốc đồng 24
thời làm tăng nồng độ ở trạng thái ổn định trong huyết tương của cả hai thuốc. Ngược lại,
metronidazol là chất ức chế không cạnh tranh các enzym ở dịch bào tương và ức chế
chuyển hóa của phenytoin, warfarin và sulphonylurea (như glyburide).
Nghiên cứu về vai trò của các isoenzym khác nhau của CYP 450 trong chuyển hóa
của một thuốc có thể tiến hành in vitro (bên ngoài cơ thể), điều này giúp dự đoán các
tương tác thuốc có thể xảy ra. Tuy nhiên, những tương tác nghiên cứu in vitro này không
phải luôn luôn có ý nghĩa trên lâm sàng trên một bệnh nhân cụ thể vì có sự khác nhau
giữa các cá nhân liên quan đến hoạt tính enzym gan, điều này dẫn đến hậu quả lâm sàng
của tương tác thuốc liên quan đến CYP 450 có thể xảy ra ở bệnh nhân này mà không có ý
nghĩa ở bệnh nhân khác.
Bảng 3.1. Một số chất cảm ứng và chất ức chế enzym Enzym
Cơ chất chuyển hóa Chất ức chế Chất cảm ứng CYP1A2 Caffeine, Clozapine, Amiodarone, Cimetidine, Insulin, Theophyllin, Warfarin Fluoroquinolone, Nafcillin, (R) Fluvoxamine Omeprazole CYP2C9a Celecoxib, Losartan, Amirone, Fluconazole, Bartiturate, NSAID, Sulphonylure, Fluoxetine/Fluovoxamine, Rifampicin Phenytoin, Warfarin (S) Lansoprazole, Sulfamethoxazole, Ticlopidine CYP2C19a Diazepam, Fluoxetine, Ketoconazole Carbamazepine, Moclobamide, Prednisone, Omeprazole, Rifampicin Pantoprazole, Proguanil CYP2D6a Codeine, Amiodarone, Celecoxib, Dexamethasone, Dextromethorphan, Cimetidine, Fluoxetine, Rifampicin Haloperidol, Quinidine Metoprolol, Nortriptyline, Pravastatin, Propanfenone CYP2E1 Chlormezanone, Diethyldithio-carbamate Ethanol, Paracetamol, Isoniazid Theophyllin CYP3A4 Alprazolam, Gestodene, Fluvoxamine, Barbiturate, Atorvastatin, Fluconazole/traconazole, Carbamazepine, 25 Ciclosporin, Ketoconazole, Efavirenz, Hydrocortisone, Nefazodone, Glucocorticoid , Lidocaine, Lovastatin, Nelfinavir/Ritonavir, và các sterioid Midazolam, Nifedipine, Verapamil, Voriconazole khác, Tamoxifen, Vincristine Nevirapine, Phenytoin, Pioglitazone
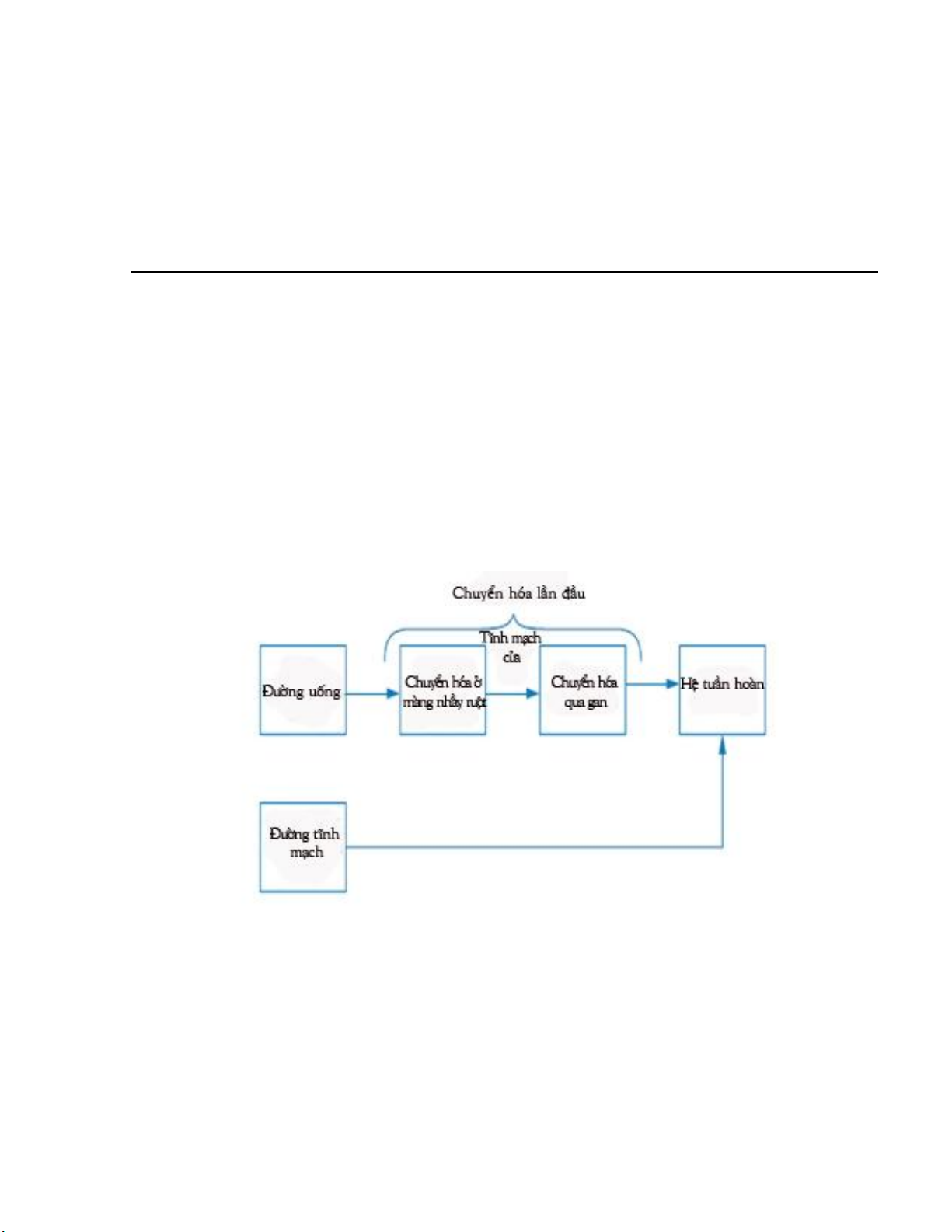
4. Chuyển hóa qua gan lần đầu (First-pass effect)
Chuyển hóa của một thuốc phụ thuộc nhiều vào đường đưa thuốc. Sau khi dùng
đường uống, đầu tiên, phân tử thuốc tiếp xúc với niêm mạc đường tiêu hóa, sau đó thuốc
được hấp thu và qua tĩnh mạch cửa vào gan trước khi vào hệ tuần hoàn và phân bố đến
phần còn lại của cơ thể. Vì vậy, nếu một thuốc bị chuyển hóa tại nhung mao ruột và/hoặc
một thuốc có độ thanh thải qua gan cao (ví dụ như thuốc đó bị chuyển hóa nhanh qua
gan), thì một phần lớn thuốc sẽ bị chuyển hóa trước khi thuốc có thể vào được hệ tuần
hoàn. Quá trình chuyển hóa này được gọi là chuyển hóa qua gan lần đầu (Hình 3.1).
Hình 3.1. Chuyển hóa qua gan lần đầu
Vì vậy, đường đưa thuốc ảnh hưởng đến quá trình chuyển hóa qua gan lần đầu. Ví
dụ, khi salbutamol được dùng ở bệnh nhân hen, tỉ lệ thuốc ở dạng không đổi/chất chuyển
hóa trong nước tiểu là 2:1 sau khi dùng đường tĩnh mạch, nhưng tỷ lệ này là 1:2 sau khi
dùng đường uống. Propranolol chuyển hóa qua gan lần đầu mạnh, vì vậy nếu dùng một 26
liều nhỏ thuốc đường uống, lượng thuốc này có thể bị chuyển hóa hoàn toàn lần đầu qua
gan trước khi vào hệ tuần hoàn.
Tương tự, niêm mạc dạ dày-ruột chứa nhiều enzyme chuyển hóa thuốc như
CYP3A4, dopa-decarboxylase, catecho-O-methyl transferase (COMT), có khả năng
chuyển hóa các thuốc như ciclosporin, felodipine, levodopa, salbutamol trước khi thuốc
vào hệ tĩnh mạch cửa để vào gan.
Quá trình chuyển hóa qua gan lần đầu mạnh tại ruột (như ciclosporin, felodipine,
levodopa, salbutamol) hoặc tại gan (như felodipine, glyceryl trinitrate, morphine,
naloxone, verapamil) đòi hỏi liều đường uống cao so với đường tĩnh mạch. Vì vậy, không
bao giờ được phép ước lượng liều tiêm IV bằng liều uống thông thường của thuốc. Các
đường khác như đường trực tràng, đường dưới lưỡi, qua da…hạn chế một phần hoặc hoàn
toàn quá trình chuyển qua gan lần đầu.
Các thuốc chuyển hóa qua gan lần đầu mạnh thường sinh đáp ứng lâm sàng khác
nhau giữa các cá thể bệnh nhân. Vì những yếu tố làm thay đổi khả năng chuyển hóa
thuốc, làm thay đổi mạnh nồng độ thuốc trong máu và đây là một trong số các khó khăn
chính sử dụng thuốc này trên lâm sàng.
Dược động học của quá trình chuyển hóa qua gan lần đầu thường không tuyến tính
(ví dụ aspirin, hydralazine, propranolol) nên tăng liều thuốc không tăng tuyến tính sinh khả dụng của thuốc.
5. Chuyển hóa thuốc bởi vi khuẩn đường ruột
Quá trình chuyển hóa bởi sinh vật đường ruột là quan trọng với những thuốc trải
qua vòng tuần hoàn gan-ruột nhiều. Ví dụ, trong trường hợp của estradiol, thuốc được thải
trừ trong mật dưới dạng liên hợp với glucuronide, những enzyme sản xuất bởi vi khuẩn
làm phân hủy glucuronide, nên thuốc tự do nhiều được tái hấp thu qua đoạn cuối của hồi
tràng. Một lượng nhỏ liều thuốc (khoảng 7%) được bài tiết qua mật trong phân ở điều
kiện sinh lý bình thường. Lượng thuốc thải qua phân này sẽ tăng nếu bệnh nhân có bệnh
lý dạ dày-ruột hoặc dùng kháng sinh làm thay đổi hệ vi khuẩn đường ruột.
6. Các yếu tố ảnh hưởng đến chuyển hóa thuốc
Các yếu tố sinh - bệnh lý hoặc ngoại lai có thể làm thay đổi quá trình chuyển hóa thuốc.
6.1. Yếu tố sinh-bệnh lý
Tuổi: chuyển hóa thuốc qua gan chưa phát triển hoàn thiện ở trẻ sơ sinh (trẻ dưới
40 tuần tuổi thai) và tiếp tục hoàn thiện đến mức ổn định khi trẻ được 3-6 tháng
tuổi. Ở trẻ sinh thiếu tháng (< 35 tuần ), chức năng chuyển hóa thuốc ở gan cần
nhiều thời gian hơn để được hoàn thiện. Do đó, trẻ sơ sinh thiếu nhiều enzyme 27
chuyển hóa thuốc. Ở người lớn tuổi, enzyme bị lão hóa. Quá trình chuyển hóa
thuốc trên cơ thể trẻ em thường nhanh hơn người lớn, tính trên cơ sở trọng lượng
cơ thể. Tốc độ chuyển hóa chậm dần cho tới tuổi trưởng thành. Ở bệnh nhân lớn
tuổi (trên 65 tuổi), độ thanh thải qua gan của một số thuốc có thể giảm, tuy nhiên
do tình trạng bệnh làm thay đổi tác dụng dược lý của thuốc nên sự ảnh hưởng do
tuổi cao bị lu mờ. Ở người cao tuổi, khối lượng gan giảm, chức năng chuyển hóa
thuốc của các tế bào gan còn lại cũng giảm.
Đặc điểm chủng tộc, sự khác biệt về gen, di truyền: luôn có sự khác nhau nhất
định về chuyển hóa giữa các cá nhân, chủng tộc và khi có sự khác nhau về kiểu
gen. Hiện tượng khác nhau về kiểu gen dẫn đến khác nhau về kiểu hình trong
trường hợp này là sự khác nhau về khả năng chuyển hóa, hiện tượng này được gọi
là hiện tượng đa hình (polymorphism). Sự khác nhau về kiểu gen có thể làm xuất
hiện các enzyme không điển hình hay thiếu enzyme tham gia chuyển hóa thuốc. Ví
dụ, có thể phân biệt mọi người thành hai nhóm đối với quá trình chuyển hóa
isoniazide: Nhóm những người chuyển hóa acetyl hóa nhanh và nhóm người
chuyển hóa acetyl hóa chậm. Thông qua các xét nghiệm về gen có thể biết trước
một cá nhân thuộc nhóm nào để từ đó điều chỉnh liều isoniazide cho phù hợp vì
isoniazide chuyển hóa bằng phản ứng acetyl hóa. Hiện tượng đa hình gen còn biểu
hiện với CYP2D6, CYP1C9, CYP2C19.
Suy gan: suy gan gây giảm chuyển hóa thuốc (như xơ gan, viêm gan mạn, viêm
gan virus). Vì vậy trong một số trường hợp, cần phải điều chỉnh liều thuốc.
6.2. Yếu tố ngoại lai
Thức ăn: khả năng chuyển hóa của gan có thể tăng (gọi là hiện tượng “cảm
ứng enzyme”) dưới ảnh hưởng của một số loại thức ăn như thịt nướng trên than,
bắp cải, bông cải xanh… Những thức ăn này, cũng giống như khói của thuốc lá, có
chứa các chất gây cảm ứng enzyme làm thúc đẩy quá trình chuyển hóa của một số thuốc như theophyllin.
Rượu: ảnh hưởng của rượu có thể chia thành hai giai đoạn: đối với người thỉnh
thoảng uống, hấp thu rượu cấp gây ức chế quá trình chuyển hóa của một số thuốc.
Ngược lại, uống rượu nhiều lần và lạm dụng rượu (nghiện rượu) gây cảm ứng
enzyme làm tăng quá trình chuyển hóa thuốc.
Thuốc dùng kèm: quá trình chuyển hóa qua gan của một thuốc có thể bị thay đổi
do dùng đồng thời với một thuốc khác, gây ức chế hay cảm ứng enzyme gan gọi là
tương tác thuốc-thuốc. Ví dụ rifampicin để điều trị bệnh lao có thể làm cho thuốc
tránh thai đường uống không còn hiệu quả do rifampicine làm cảm ứng enzyme
làm tăng chuyển hóa thuốc tránh thai. 28
Câu hỏi lượng giá:
1. Tóm tắt các giai đoạn chính của chuyến hóa thuốc.
2. Phân tích ảnh hưởng của hiện tượng cảm ứng, ức chế enzyme cytochrome P450 lên
quá trình chuyển hóa của thuốc.
3. Trình bày quá trình chuyển hóa thuốc qua gan lần đầu và chuyển hóa thuốc do vi khuẩn ruột.
4. Phân tích các yếu tố ảnh hưởng đến chuyển hóa thuốc. Ca lâm sàng
Một phụ nữ 46 tuổi nhập viện vào khoa cấp cứu, do uống một số lượng không rõ
viên nén paracetamol bằng rượu vodka sáu giờ trước, sau khi cải vã với chồng. Bệnh nhân
là một người nghiện rượu mạn và bệnh nhân đang dùng carbamazepine liều 200mg/ngày,
chia hai lần đã hơn 1 tháng để điều trị đau thần kinh. Ngoài các dấu hiệu ngộ độc, các
thăm khác chức năng khác của cơ thể bình thường. Nồng độ paracetamol trong huyết
tương là 662 µmol/l (100mg/l). Câu hỏi:
1. Yếu tố nguy cơ làm tăng khả năng quá liều paracetamol trên bệnh nhân này không?
Biết nghiện rượu, carbamazepine có khả năng cảm ứng enzyme CYP1A2, CYP3A4 và các CYP khác.
2. Thuốc khử độc đặc hiệu trong trường hợp quá liều paracetamol là gì? Cơ chế? Trình
bày khái niệm và đặc điểm của quá trình chuyển hóa thuốc. 29
BÀI 4: THẢI TRỪ THUỐC MỤC TIÊU
1. Trình bày được quá trình thải trừ thuốc qua thận, qua mật.
2. Phân tích được các yếu tổ ảnh hưởng đến quá trình thải trừ thuốc.
3. Trình bày các thông số độ thanh thải của thuốc, thời gian bán thải của thuốc và ý nghĩa của nó. NỘI DUNG
1.Khái niệm thải trừ thuốc
Thải trừ thuốc là quá trình loại bỏ thuốc ra khỏi cơ thể được thực hiện thông qua
nhiều đường khác nhau, chủ yếu qua thận, mật; ngoài ra còn được đào thải qua đường qua
phổi, da, lông tóc, mồ hôi, nước mắt và nước bọt. Thải trừ thuốc là con đường giải độc của cơ thể.
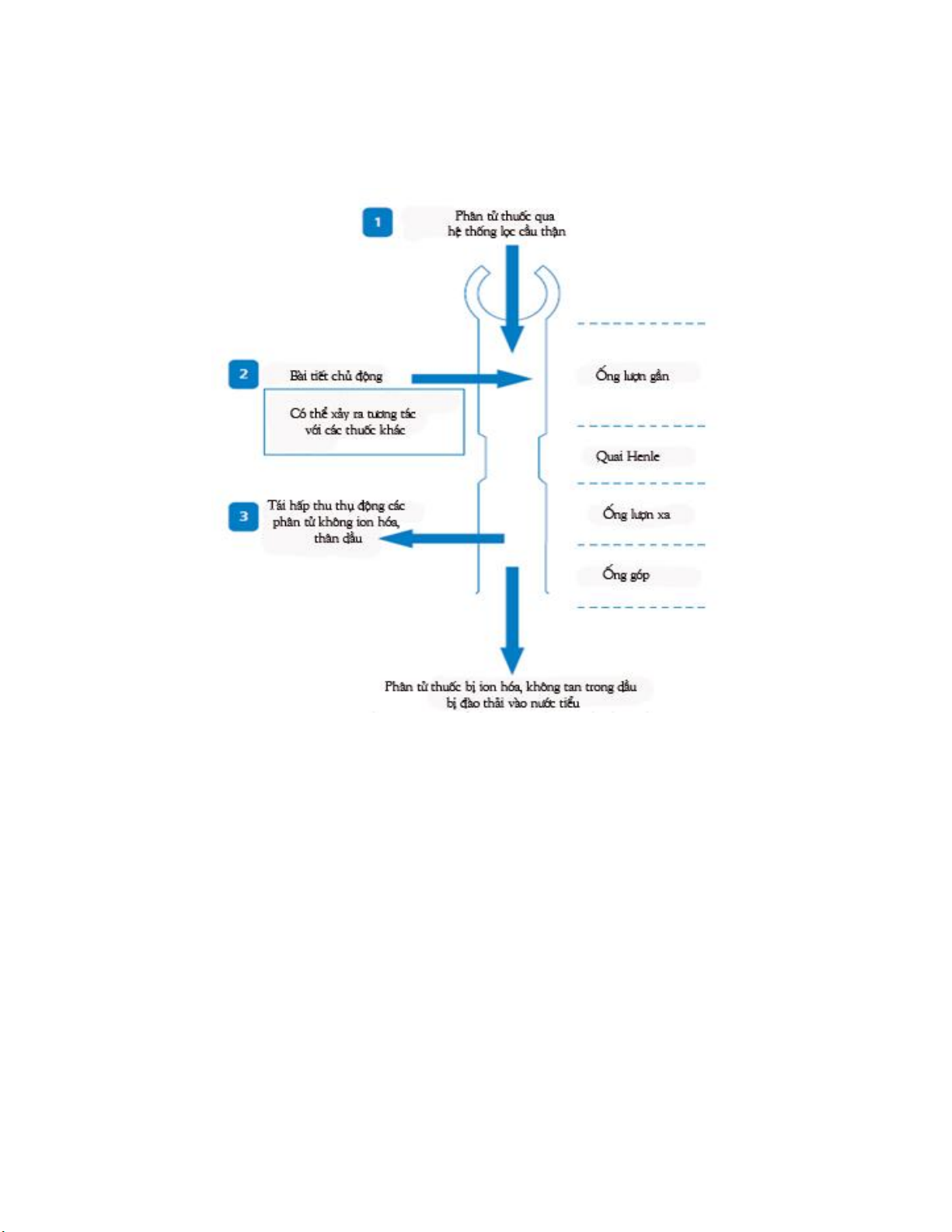
2. Thải trừ thuốc qua thận
Thận là cơ quan thải trừ thuốc chính ở trong cơ thể (Hình 4.1). Qua quá trình
chuyển hóa, thuốc được chuyển hóa thành các chất dễ tan trong nước hơn, sau đó được
đào thải qua thận dễ dàng. Các chất thân nước dễ dàng được đào thải qua thận vì nó
không thể khuếch tán tự do qua màng của ống thận để trở lại dịch kẽ mà bị giữ lại trong
nước tiểu dù nồng độ chất đó trong nước tiểu rất cao.
Lọc ở cầu thận: Cầu thận là đơn vị chức năng của thận có chức năng đào thải các
chất thải đồng thời đào thải các phân tử thuốc ra khỏi cơ thể. Các phân tử lớn hơn hoặc
bằng 66 000 kDa (bao gồm các protein huyết tương, các phức thuốc-protein) không thể
qua được màng lọc cầu thận. Chỉ các phân tử thuốc tự do có kích thước tương đối nhỏ và
được lọc qua cầu thận theo cơ chế vận chuyển thụ động. Quá trình lọc qua cầu thận là con
đường thải trừ chính của nhiều loại thuốc. Những thuốc được thải trừ chủ yếu qua quá
trình lọc ở cầu thận, trong trường hợp bệnh nhân bị tổn thương thận gây giảm tốc độ lọc
cầu thận, làm giảm thải trừ thuốc như digoxin.
Bài tiết chủ động ở ống lượn gần: Một số thuốc có thể được bài tiết chủ động vào
nước tiểu ở ống lượn gần. Quá trình bài tiết này là chủ động nhờ vào các chất mang hoặc
các bơm, vận chuyển thuốc dưới dạng ion hóa (ion âm, ion dương) từ các mạch máu gần
với cầu thận vào phần ống lượn gần của thận. Các thuốc bài tiết theo cơ chế này như
probenecid, penicilin, cimetidine, amphetamine.
Đây là nơi chính xảy ra tương tác thuốc tại thận do sự cạnh tranh chất mang hoặc
bơm vận chuyển. Ví dụ, probenecid ức chế hoàn toàn sự bài tiết của methotrexate ở ống
thận làm tăng nồng độ methotrexate trong máu. 30
Para-aminohippuric acid (PAH) được tiết rất mạnh qua cơ chế này đến nỗi toàn bộ
PAH được loại ra khỏi huyết tương thận sau khi chỉ qua thận một lần. Vì vậy, độ thanh
thải của PAH chính là thông số biểu thị dòng huyết tương qua thận.
Hình 4.1. Quá trình bài tiết và tái hấp thu thuốc qua thận
Tái hấp thu ở ống thận: Song song với quá trình đào thải, một số loại thuốc có
thể được tái hấp thu thụ động tại ống lượn xa hoặc tái hấp thu chủ động ở ống lượn gần từ
nước tiểu trở lại vào máu qua thận.
Quá trình tái hấp thu thụ động ở ống lượn xa đòi hỏi phân tử thuốc có tính thân
lipid nhất định. Ngược lại, phân tử thân nước như mannitol không thể tái hấp thu nên hầu
như được thải trừ hoàn toàn. Vì vậy, tái hấp thu thụ động ở ống lượn xa chịu ảnh hưởng
của độ pH của nước tiểu.
Đối với tái hấp thu thuốc là acid yếu (AH) và base (B) yếu ở môi trường pH nước
tiểu thấp, thì nồng độ thuốc tồn tại nhiều ở dạng AH (thân dầu) và BH+ (thân nước). Do
đó, chủ yếu dạng AH được tái hấp thu thụ động và chủ yếu BH+ được thải trừ ra trong
nước tiểu. Ngược lại, trong môi trường pH nước tiểu cao, thì nồng độ thuốc tồn tại dạng 31
A-(thân nước) và B (thân dầu) lớn. Do đó, chủ yếu dạng B là được tái hấp thu, trong khi
chủ yếu A- được đào thải trong nước tiểu ra ngoài. Điều này được ứng dụng để xử lý quá
liều với các thuốc acid yếu và base yếu bằng cách thay đổi pH nước tiểu. Ví dụ, xử lý quá
liều aspirin (acid yếu) bằng cách kiềm hóa nước tiểu, vì vậy tăng thải trừ aspirin.
Tái hấp thu ở ống thận còn phụ thuộc vào tốc độ dòng nước tiểu. Tái hấp thu nhiều
ở tốc độ dòng nước tiểu chậm và ngược lại. Vì vậy, thuốc lợi tiểu được dùng để xử lý ngộ độc thuốc.
Quá trình tái hấp thu chủ động ở ống lượn gần: Thường ít quan trọng đối với hầu
hết các thuốc. Acid uric được tái hấp thu bởi hệ thống chất mang mà hệ thống này lại bị
ức chế bởi các thuốc như probenecid, sulfinpyrazone, do đó các thuốc này làm tăng acid
uric niệu. Lithium cũng được tái hấp thu chủ động ở đây.
3. Thải trừ thuốc qua mật
Sau khi qua gan, phần thuốc không được chuyển hóa có thể quay lại hệ tuần hoàn
chung hoặc được thải trừ qua mật. Thuốc được thải trừ qua mật sẽ trải qua vòng tuần hoàn
ruột-gan rồi được đổ vào túi mật, sau đó vào lại trong ruột và có thể được hấp thu tiếp qua
màng ruột để vào máu và quay trở lại gan. Thải trừ qua mật thông thường là một đường
thải trừ phụ, nhưng nó có thể trở thành con đường chính khi các đường thải trừ khác như thận giảm hoạt động.
4. Các yếu tố ảnh hưởng đến thải trừ thuốc
Các yếu tố sinh-bệnh lý hoặc ngoại lai có thể làm thay đổi quá trình thải trừ thuốc.
4.1. Yếu tố sinh-bệnh lý
Suy thận có thể làm giảm thải trừ thuốc và gây tích lũy thuốc. Giảm thải trừ thuốc
tỉ lệ với mức độ suy thận. Mức độ suy thận có thể được đánh giá thông qua một vài xét
nghiệm sinh hóa như creatinine, độ thanh thải của creatinin. Liều hoặc số lần đưa
thuốc/ngày của một số thuốc được điều chỉnh dựa trên mức độ suy thận. Tất cả những
thay đổi về mức độ gắn với protein huyết tương của thuốc làm thay đổi phần thuốc dạng
tự do, thay đổi thải trừ thuốc.
4.2. Yếu tố ngoại lai
Tất cả thay đổi về pH nước tiểu rất dễ làm thay đổi quá trình tái hấp thu thụ động ở
ống thận, vì nó làm thay đổi phần thuốc không ion hóa của thuốc. Ví dụ, kiềm hóa nước
tiểu làm tăng ion hóa các thuốc acid yếu (như phenobarbital), làm giảm đáng kể tái hấp
thu của chúng. Vì vậy, kiềm hóa nước tiểu thường bằng truyền dung dịch natri
bicarbonate là một xử lý phổ biến để giải độc phenobarbital.
Có thể xảy ra hiện tượng cạnh tranh giữa các thuốc trong quá trình bài tiết ở ống
thận như cạnh tranh giữa thuốc hạ đường huyết sulfamide và phenylbutazone làm giảm 32
bài tiết sulfamide vào nước tiểu, làm giảm thải trừ thuốc, nên nồng độ thuốc trong máu
cao, dẫn đến làm hạ đường huyết quá mức nghiêm trọng ở bệnh nhân.
5. Độ thanh thải của thuốc và thời gian bán thải thuốc
5.1. Độ thanh thải của thuốc (Clearance-Cl)
Độ thanh thải (Clearance, kí hiệu là Cl) biểu thị khả năng của một cơ quan nào đó
của cơ thể (thường là gan và thận) lọc sạch thuốc ra khỏi huyết tương khi máu tuần hoàn
qua cơ quan đó. Cụ thể, nó được tính bằng thể tích huyết tương được lọc sạch thuốc sau
khi qua cơ quan đó trong một đơn vị thời gian.
Đơn vị của độ thanh thải Cl là l/giờ hay ml/phút.
Độ thanh thải Cl là một giá trị ảo, có tính lý thuyết vì sự tuần hoàn của máu qua
các cơ quan là hồi lưu, lặp đi lặp lại liên tục.
Độ thanh thải toàn phần là tổng độ thanh thải của toàn bộ các cơ quan trong cơ thể
tham gia đào thải thuốc:
Cltoàn phần = Cl thận + Clgan + Cl cơ quan khác ≈ Cl thận + Clgan
Thông thường, thuốc chuyển hóa chủ yếu ở gan thì tổn thương thận ít có nguy cơ
gây độc (ví dụ theophylin chuyển hóa 90% qua gan). Trong khi đó, thuốc chủ yếu bài
xuất qua thận nếu được sử dụng ở bệnh nhân có tổn thương thận sẽ làm khả năng bài xuất
thuốc giảm gây nguy cơ quá liều cao (ví dụ cephalexin đào thải qua thận 91%).
5.2. Thời gian bán thải của thuốc
Thời gian bán thải của thuốc hay còn gọi là thời gian bán thải (t ) là thời gian cần 1/2
thiết để thải trừ một nửa nồng độ thuốc trong máu.
Ví dụ, nếu nồng độ thuốc trong huyết tương của phenytoin (thuốc chống động
kinh) là 20µg/ml sau khi dùng một liều thuốc duy nhất và nếu thời gian cần thiết để giảm
nồng độ xuống còn 10µg/ml là 24 giờ, thì thời gian bán thải trong trường hợp này là 24 giờ.
Giá trị thời gian bán thải thuốc phụ thuộc vào quá trình chuyển hóa và thải trừ
thuốc. Thời gian bán thải của một thuốc thay đổi giữa người này so với người khác phụ
thuộc vào tình trạng sinh – bệnh lý của từng cá nhân.
Thời gian bán thải là một thông số dược động học quan trọng, bởi vì nó quyết định
số lần dùng thuốc trong ngày. Thuốc có thời gian bán thải càng ngắn thì càng cần phải
dùng nhiều lần trong ngày.
Khi dùng thuốc đường uống lâu dài, thuốc tích lũy trong cơ quan cho đến khi tốc
độ thải trừ bằng tốc độ hấp thu thuốc. Trạng thái cân bằng là trạng thái tại đó nồng độ 33
thuốc ổn định, không tăng thêm được nữa. Trạng thái cân bằng thường đạt được sau
khoảng 5 đến 6 lần thời gian bán thải thuốc. Ví dụ, thời gian bán thải của phenytoin là 24
giờ, trạng thái cân bằng đạt được sau 5-6 ngày.
Tuy nhiên, thời gian bán thải không là thông số trực tiếp đo lường quá trình thải trừ
thuốc bởi vì t1/2 thay đổi có thể do thay đổi ở hiệu quả thải trừ (Cl) hoặc thay đổi thể tích
phân bố (Vd). Chính độ thanh thải (Cl) mới chính là thông số trực tiếp đo lường hiệu quả
của quá trình thải trừ thuốc.
Câu hỏi lượng giá
1. Trình bày quá trình thải trừ thuốc qua thận, qua mật.
2. Phân tích các yếu tổ ảnh hưởng đến quá trình thải trừ thuốc.
3. Trình bày các thông số độ thanh thải của thuốc, thời gian bán thải của thuốc và ý nghĩa của nó. 34
BÀI 5: CÁC MÔ HÌNH DƯỢC ĐỘNG HỌC CƠ BẢN MỤC TIÊU 1.
Phân tích được sự khác nhau giữa mô hình dược động học một ngăn và mô hình
dược động học hai ngăn. 2.
Ứng dụng tính toán các thông số dược động học cơ bản, khoảng cách đưa thuốc,
liều tấn công, liều duy trì đối với 3 cách đưa thuốc. NỘI DUNG
Trong các chương trước, chúng ta đã miêu tả về quá trình hấp thu, phân bố, chuyển
hóa, thải trừ riêng rẻ của thuốc trong cơ thể. Ở bài này, chúng ta sẽ dùng các mô hình định
lượng đơn giản, các khái niệm toán học để miêu tả tất cả các quá trình trên diễn ra đồng thời như thế nào.
Hiểu những nguyên tắc cơ bản của dược động học, kết hợp với các thông tin cụ thể
liên quan đến thuốc và bệnh nhân, giúp sử dụng thuốc tối ưu cho từng bệnh nhân như
chọn thuốc, đường dùng, liều lượng và khoảng cách liều.
Dược động học bao gồm các nghiên cứu nồng độ thuốc (trong máu, nước tiểu, mô)
theo thời gian, giúp xác định các giai đoạn khác nhau của thuốc trong cơ thể (hấp thu,
phân bố chuyển hóa, thải trừ). Có thể chia dược động học làm hai mảng: dược động học
cơ bản và dược động học lâm sàng. Tuy nhiên, cả hai mảng này đều tuân theo cùng những quy luật giống nhau.
Dược động học cơ bản là nghiên cứu dược động học tiến hành của thuốc tiến hành
ở động vật hoặc người bình thường (pha I của nghiên cứu lâm sàng) trong điều kiện sinh
lý tối ưu, giúp xác định các thông số dược động học cũng như tìm hiểu sự khác nhau của
thông số dược động học khi dùng các liều khác nhau.
Dược động học lâm sàng nghiên cứu dược động học của thuốc ở bệnh nhân, có thể
tiến hành trong quá trình phát triển thuốc (pha II, III) hoặc sau khi thuốc được lưu hành
trên thị trường. Các nghiên cứu này giúp xác định sự khác nhau về các thông số dược
động học trên các bệnh lý, các phối hợp điều trị, các cách dùng thuốc khác nhau… Mục
đích của dược động học là tính toán liều và khoảng cách đưa thuốc để nồng độ thuốc đạt
tối ưu trên bệnh nhân hoặc giúp điều chỉnh liều nếu lựa chọn liều ban đầu không thích
hợp. Vì vậy, dược động học là một công cụ quan trọng để theo dõi điều trị bệnh nhân.
Mô hình dược động học được đưa ra dựa trên những giả thiết giúp đơn giản hóa
việc tính toán. Trong chương này chỉ giới thiệu 3 trường hợp đưa liều trên lâm sàng:
truyền tĩnh mạch với tốc độ không đổi, tiêm bolus đơn liều và dùng liều bolus lặp lại. 35
1. Mô hình một ngăn với quá trình thải trừ bậc 1
Mô hình dược động học đơn giản nhất xem cơ thể như một ngăn đơn luôn được
khuấy trộn đều, trong đó thuốc sau khi dùng được phân bố tức thì và từ đó thuốc được
thải trừ. Nhiều thuốc được thải trừ với tốc độ tỷ lệ với nồng độ của chúng, gọi là thải trừ
“bậc 1”. Mô hình một ngăn đơn với thải trừ bậc 1 thường mô tả gần đúng với tình huống
trên lâm sàng khi giai đoạn hấp thu và phân bố đã hoàn thành và giai đoạn thải trừ thuốc bắt đầu.
1.1. Truyền thuốc với tốc độ không đổi
Nếu một thuốc được dùng đường tĩnh mạch bằng bơm với tốc độ không đổi và lấy
mẫu máu tĩnh mạch để đo lường nồng độ thuốc trong huyết tương (Cp) sau các khoảng
thời gian t, thu được một đồ thị biểu diễn nồng độ thuốc trong huyết tương theo thời gian như Hình 5.1.
Nồng độ tăng nhanh dần từ 0, sau đó tăng chậm dần cho đến khi nồng độ ổn định
được thiết lập (Css). Trạng thái tại đó nồng độ thuốc ổn định gọi là trạng thái ổn định –
steady state). Ở trạng thái ổn định, tốc độ hấp thu thuốc vào cơ thể bằng tốc độ thải trừ
thuốc ra khỏi cơ thể (Công thức 1). Trong đó, độ thanh thải (Clearance) được định nghĩa
là thể tích dịch (thường là huyết tương) từ đó thuốc được lọc sạch thuốc trong một đơn vị thời gian (Công thức 2).
Ở trạng thái cân bằng:
Tốc độ truyền thuốc = Tốc độ thải trừ thuốc (1) Do đó:
Theo công thức 3 cho thấy, nồng độ ổn định Css phụ thuộc vào tốc độ truyền thuốc và độ
thanh thải (Cl) của thuốc. 36
Hình 5.1. Nồng độ thuốc trong huyết tương theo thời gian trong và sau khi
truyền tĩnh mạch với tốc độ không đổi
Độ thanh thải (kí hiệu Cl) là thông số tốt nhất đo lường hiệu quả đào thải thuốc ra
khỏi cơ thể, qua con đường chuyển hóa, hay thải trừ qua thận hoặc cả hai. Khái niệm này
tương tự với khái niệm độ thanh thải của các chất đặc biệt được dùng như thông số đánh
giá các quá trình sinh lý quan trọng của cơ thể như tốc độ lọc cầu thận, dòng huyết tương qua gan hay qua thận.
Với các thuốc, biết được độ thanh thải của thuốc ở một bệnh nhân có thể giúp điều
chỉnh liều duy trì thuốc để đạt được nồng độ ổn định mong muốn, bởi vì:
Tốc độ truyền thuốc yêu cầu = Css mong muốn x Cl (4)
Thông tin này hữu ích trong quá trình nghiên cứu, phát triển thuốc đồng thời hữu
ích trên lâm sàng khi điều trị một số thuốc dựa trên nồng độ thuốc đo lường trong huyết tương.
Khi dừng truyền thuốc, nồng độ thuốc trong huyết tương dần giảm về 0. Thời gian
để nồng độ thuốc trong huyết tương giảm còn 1/2 được gọi là thời gian bán thải (t1/2).
Mô hình một ngăn với quá trình thải trừ bậc 1 có phương trình nồng độ thuốc trong
huyết tương giảm theo thời gian tuân theo hàm số mũ khi dừng truyền thuốc, như Hình 5.1.
Nồng độ thuốc trong huyết tương tăng dần theo thời gian cũng tuân theo hàm số
mũ khi bắt đầu truyền thuốc, nhưng theo hướng nghịch đảo với hàm số mũ khi thải trừ.
Điều này rất có ý nghĩa trên lâm sàng, vì điều đó cho thấy t1/2 không chỉ giúp xác định
nồng độ thuốc - thời gian khi thuốc dần được thải trừ khi dừng truyền thuốc, mà còn giúp
xác định nồng độ thuốc-thời gian trong quá trình tích lũy thuốc để đạt trạng thái ổn định
từ khi bắt đầu truyền thuốc. 37
1.2. Tiêm bolus một liều đơn
Tiêm bolus một liều đơn là tiêm nhanh một lượng lớn thuốc vào tĩnh mạch nhằm
làm tăng nhanh nồng độ thuốc trong máu đến nồng độ điều trị yêu cầu.
Thể tích phân bố Vd được định nghĩa là thể tích giả kiến để phân phối một liều
thuốc D tiêm bolus vào tĩnh mạch để đạt được nồng độ thuốc trong huyết tương là Cp. Do đó: Vd = D/Cp
Tuy nhiên, trong thực tế, ngay khi liều bolus (D) được tiêm thì quá trình thải trừ
thuốc đã bắt đầu ngay với độ thanh thải Cls (độ thanh thải hệ thống). Trong thực hành,
máu được lấy mẫu sau những quãng thời gian gần nhau sau khi tiêm thuốc để xác định đồ
thị nồng độ-thời gian, từ đó tính được AUC. Từ hai giá trị đã biết này, tính được độ thanh thải hệ thống: Cls = D/AUC (5)
Trong trường hợp dùng đường khác (ví dụ đường uống) với sinh khả dụng F, thì
độ thanh thải hệ thống được tính như sau: Cls =( F×D)/AUC (5’)
Nếu là mô hình một ngăn, quá trình thải trừ bậc 1 thì đồ thị nồng độ thuốc giảm
theo thời gian tuân theo hàm số mũ giống như giai đoạn cuối của truyền thuốc với tốc độ
không đổi (Hình 5.1). Nếu chuyển sang hàm logarit của nồng độ thì đồ thị sẽ là một
đường thẳng có hệ số góc âm.
Hình 5.2. Mô hình một ngăn sau khi tiêm bolus liều đơn. Đường cong nồng
độ thuốc-thời gian tuân theo hàm số mũ (a) và đồ thị bán logarit
hóa nồng độ thuốc tuyến tính (b)
Từ đồ thị này, ngoại suy về thời điểm t , sẽ tìm ra được giá trị C 0 0 và Vd được tính như sau: 38 Vd = D/C0 (6)
Thời gian bán thải t có thể được tính trên đồ thị bằng cách dựa vào khoảng thời gian cần 1/2
thiết từ nồng độ C1 bất kì đến nồng độ 1/2C1.
Hằng số tốc độ thải trừ ke chính là hệ số gốc của đồ thị logarit (nồng độ)-thời gian và
được tính theo công thức: ke = Cls/Vd (7)
Hằng số tốc độ thải trừ kel chính là hệ số góc của đường thẳng biểu diễn logarit nồng độ
(lnC) theo thời gian t, nên nó có thể được tính toán từ đồ thị như sau: (7’)
t1/2 và ke liên hệ với nhau qua công thức:
t1/2 = ln2/ke = 0,693/ke (8)
Vd phụ thuộc một phần vào đặc điểm của thuốc (ví dụ khả năng thân dầu), một
phần vào đặc điểm của bệnh nhân (ví dụ kích thước cơ thể, nồng độ protein huyết tương,
khối lượng nước, mỡ). Nói chung, các chất thân dầu cao có thể xâm nhập vào tế bào và
mô mỡ nên có thể tích phân bố Vd lớn hơn các chất thân nước.
Theo công thức (6) cho thấy Vd quyết định nồng độ đỉnh (C ) trong huyết tương 0
sau khi tiêm liều bolus D, vì vậy những yếu tố làm thay đổi Vd (ví dụ khối lượng cơ thể,
phân bố của cơ thể khác nhau giữa người trưởng thành và trẻ sơ sinh hoặc người già) cần
được xem xét khi quyết định liều D (ví dụ tính liều theo kg cân nặng). 39
Biết Vd còn hữu ích khi xem xét thẩm tách máu để thải trừ thuốc ở bệnh nhân bị
ngộ độc thuốc. Thuốc với Vd lớn (ví dụ các thuốc chống trầm cảm ba vòng) không bị loại
bỏ hiệu quả sau khi thẩm tách máu bởi vì chỉ một lượng nhỏ toàn bộ lượng thuốc trong cơ
thể là nằm trong huyết tương, trong khi quá trình thẩm tách chỉ có thể tách thuốc ra khỏi
máu chứ không thể tách thuốc ra khỏi mô, cơ quan.
Nếu cả Vd và t1/2 đều được biết, chúng có thể được dùng để ước lượng độ thanh
thải hệ thống của thuốc: (9)
Chú ý rằng độ thanh thải được tính bằng đơn vị thể tích/thời gian (ví dụ ml/phút),
Vd có đơn vị thể tích (ví dụ ml hay l), t1/2 có đơn vị thời gian (ví dụ phút) và 0,693 là hằng số.
Ngoài ra, độ thanh thải thuốc còn có thể tính dựa theo nồng độ thuốc thải trừ trong
nước tiểu theo công thức: Trong đó:
Cl: Độ thanh thải của thuốc (ml/ph;l/giờ)
Cu: Nồng độ thuốc còn hoạt tính trong nước tiểu(mg/ml; g/l)
Vu: Thể tích nước tiểu (ml/ph; l/giờ)
Cp: Nồng độ thuốc còn hoạt tính trong huyết tương (mg/ml;g/l)
Công thức này cho thấy mối liên hệ giữa Cls với Vd và t , nhưng không giống với 1/2
công thức (3) ở trạng thái cân bằng khi truyền thuốc với tốc độ không đổi hoặc công thức
(5) dùng phương pháp tính AUC sau khi tiêm bolus hoặc công thức (10) đúng trong mọi
trường hợp, trong khi công thức (9) chỉ áp dụng đối với thuốc tuân theo mô hình dược
động học một ngăn có quá trình thải trừ bậc 1.
1.3. Tiêm liều bolus lặp lại
Nếu liều thuốc được tiêm nhiều lần với các khoảng cách đưa liều là lớn hơn nhiều
so với thời gian bán thải của thuốc thì quá trình tích lũy thuốc hầu như không xảy ra
(Hình 5.3 (a)). Thuốc thỉnh thoảng được cho liều như thế này (ví dụ penicillin để điều trị
một nhiễm trùng nhẹ), nhưng nồng độ ở trạng thái cân bằng cần lớn hơn một giá trị
ngưỡng để sinh đáp ứng ổn định trong suốt quãng thời gian dùng thuốc. Hình 5.3 (b) cho 40
thấy đường cong nồng độ-thời gian khi tiêm bolus lặp lại ở khoảng cách đưa thuốc nhỏ
hơn t . Quá trình tích lũy thuốc vì thế xảy ra. Nồng độ trung bình tăng cho đến khi đạt 1/2
đến bình nguyên (plateau) tương tự như khi thuốc được truyền với tốc độ không đổi.
Nghĩa là, sau t1/2 thời gian thì nồng độ trung bình bằng 50% nồng độ ổn định trung bình Css, sau 2t thời gian thì đạt 87,
1/2 thời gian thì đạt 75%; sau 3t1/2 5% và sau 4t1/2 thời gian
thì nồng độ trung bình thuốc trong huyết tương đạt 93,8% nồng độ trung bình ổn định.
Hình 5.3. Tiêm liều bolus lặp lại (mỗi mũi tên biểu thị một lần tiêm) với (a):
khoảng cách liều lớn hơn t1/2 và (b): khoảng cách liều nhỏ hơn t1/2.
Liên quan giữa khoảng cách đưa thuốc và t1/2
Liên quan giữa khoảng cách đưa liều (τ) và thời gian bán thải (t ) được thể hiện qua 1/2 công thức Co ln Ct x t (11) 1/ 2 0, 693 41
Trong đó τ là khoảng cách đưa liều để đưa thuốc từ nồng độ thuốc hiện tại (C ) đến 0
nồng độ mong muốn (Ct).
Tuy nhiên, không giống truyền thuốc với tốc độ không đổi, nồng độ thực sự của
thuốc trong huyết tương tại thời điểm bất kì dao động trên hoặc dao dộng dưới xung
quanh giá trị nồng độ trung bình. Tăng số lần đưa thuốc (khoảng cách đưa thuốc gần
nhau) sẽ làm nồng độ đỉnh (Cmax) và nồng độ đáy (Cmin) gần nhau hơn. Nếu nồng độ
đỉnh quá cao, độc tính sẽ xảy ra, trong khi nếu nồng độ đáy quá thấp sẽ làm mất tác dụng
điều trị của thuốc. Nếu một thuốc được dùng một lần mỗi t thì theo công thức (11) 1/2
Cmax sẽ gấp đôi Cmin. Trong thực hành, sự khác nhau Cmax = 2 Cmin là có thể chấp
nhận được trong nhiều trường hợp lâm sàng, nên khoảng cách đưa thuốc thường là xấp xỉ t1/2.
Biết thời gian bán thải giúp bác sĩ, dược sĩ dự đoán được nồng độ theo thời gian
khi thuốc tích lũy đến trạng thái cân bằng.
Độ thanh thải thuốc, đặc biệt là độ thanh thải thận giảm theo tuổi. Một điều lưu ý
là một số thuốc bị chuyển hóa thành sản phẩm có hoạt tính và sản phẩm có hoạt tính này
lại được đào thải chậm hơn thuốc gốc (ví dụ như một số thuốc nhóm benzodiazepine có
sản phẩm chuyển hóa có t1/2 đến vài ngày). Kết quả là tác dụng không mong muốn như lú
lẫn có thể xuất hiện chỉ khi trạng thái cân bằng đạt được sau vài tuần điều trị. Những tác
dụng không mong muốn chậm này có thể được gán cho sự giảm khả năng nhận thức do
tuổi già, nhưng thực ra chỉ cần dừng thuốc tác dụng này sẽ biến mất.
Biết thời gian bán thải còn giúp bác sĩ quyết định liệu có cần bắt đầu điều trị bằng
một liều tấn công (loading dose). Xem xét trường hợp với digoxin, thời gian bán thải xấp
xỉ 40 giờ. Thuốc thường được kê một lần/ngày nên Cmax/Cmin<2 và đạt nồng độ >90%
nồng độ ổn định trung bình xấp xỉ sau 4t = 4x40 giờ = 160 giờ ≈ 7 ngày. Trong nhiều 1/2
trường hợp lâm sàng, sự thay đổi nồng độ-thời gian này có thể chấp nhận được. Tuy
nhiên, trong những tình huống cấp cứu, một liều tấn công cần được dùng để tạo đáp ứng
nhanh hơn. Liều tấn công có thể được ước lượng bằng cách nhân nồng độ thuốc trong
huyết tương mong muốn với thể tích phân bố:
Liều tấn công (Loading Dose-LD) = Cp x Vd. (12)
Nếu trong trường hợp dùng đường khác đường tĩnh mạch (như đường uống) có
sinh khả dụng F thì liều tấn công được tính như sau với S là tỷ lệ muối của thuốc (salt fraction): 42
Với mô hình dược động học một ngăn có quá trình đào thải bậc 1 thì t không phụ 1/2
thuộc nồng độ thuốc trong máu.
Khi ở trạng thái cân bằng và ngừng tiêm thuốc, thì quá trình thải trừ bậc 1 có đồ thị
nồng độ-thời gian tuân theo hàm số mũ, do đó, lượng thuốc thải trừ tỷ lệ với t1/2 như sau:
Bảng 5.1. Liên quan giữa lượng thuốc được thải trừ với t1/2 sau khi thuốc đạt trạng
thái ổn định thì dừng tiêm thuốc Số lần t
Lượng thuốc được thải trừ (%) 1/2 1 50 2 75 3 88 4 94 5 97 6 98 7 99
Như vậy, thuốc được coi bài xuất hoàn toàn khỏi cơ thể sau 7 x t . Để thay thuốc phải 1/2
chờ ít nhất 7 x t1/2 để tránh hiện tượng tích lũy.
2. Các mô hình khác với mô hình một ngăn có quá trình đào thải bậc 1
2.1. Mô hình hai ngăn
Nếu sau khi tiêm bolus tĩnh mạch, nồng độ thuốc trong huyết tương giảm theo 2
giai đoạn chứ không phải 1 giai đoạn (Hình 5.4). Mô hình hai ngăn đươc áp dụng trong
trường hợp này. Mô hình hai ngăn xem cơ thể gồm hai ngăn: ngăn trung tâm và ngăn
ngoại vi. Khái niệm ngăn này không liên quan một cách chính xác với khái niệm giải
phẫu, dù ngăn trung tâm có thể được giả định là gồm máu (từ đó mẫu máu được lấy để
phân tích) và khoảng gian bào của một số mô; còn ngăn ngoại vi bao gồm các mô mà thuốc phân bố vào. 43
Hình 5.4. Mô hình hai ngăn. Đường con
g nồng độ thuốc trong huyết tương-thời gian
sau khi tiêm một liều bolus.
Giai đoạn đầu nồng độ thuốc giảm nhanh gọi là giai đoạn alpha, thể hiện chủ yếu
quá trình thuốc phân bố từ ngăn trung tâm ra ngăn ngoại vi. Giai đoạn thứ hai nồng độ
thuốc giảm chậm hơn gọi là giai đoạn beta, thể hiện chủ yếu quá trình đào thải thuốc và
thời gian bán thải được gọi là t beta. Đây là giá trị thích hợp dùng trong lâm sàng. 1/2
2.2. Dược động học không tuyến tính (phụ thuộc liều)
Dù nhiều thuốc được thải trừ với tốc độ tỉ lệ với nồng độ thuốc (dược động học bậc
1), vẫn có vài trường hợp ngoại lệ trên lâm sàng quan trọng. Ví dụ trường hợp một thuốc
được đào thải bằng cách chuyển hóa thành chất chuyển hóa bất hoạt bởi một enzyme. Ở
nồng độ thuốc cao, enzyme chuyển hóa thuốc trở nên bảo hoà nên tốc độ thải trừ không
phụ thuộc liều thuốc nữa. Nồng độ thuốc và vận tốc phản ứng chuyển hóa liên hệ với
nhau qua công thức Michaelis-Menten (Hình 5.5).
Ở nồng độ thấp, tốc độ phản ứng chuyển hóa tỉ lệ tuyến tính với nồng độ thuốc,
trong khi ở nồng độ bảo hòa enzyme, tốc độ phản ứng chuyển hóa độc lập với nồng độ
thuốc (dược động học bậc 0).
Điều tương tự xảy ra khi một thuốc được đào thải bởi quá trình vận chuyển do một
chất mang có tính bão hòa. Trong thực hành lâm sàng, những thuốc tuân theo dược động
học bậc 0 thường những trường hợp cá biệt. Bởi vì hầu hết các thuốc được dùng điều trị
với liều thấp hơn nhiều hằng số Michaelis-Menten (Km) và vì thế, tốc độ phản ứng tỉ lệ 44
tuyến tính với nồng độ thuốc trong huyết tương (tương ứng phần phía dưới đồ thị Michaelis-Menten).
Hình 5.5. Mối liên hệ Michaelis-Menten giữa tốc độ phản ứng chuyển hóa của enzyme
(V) và nồng độ thuốc (S). Nồng độ (S) tại vị trí 50% Vmax chính là hằng số Michaelis-Menten (Km)
Các thuốc tuân theo dược động học không tuyến tính trong khoảng liều điều trị bao
gồm heparin, phenytoin và ethanol. Một số thuốc (như nhóm barbiturate) cho thấy tuân
theo mô hình không tuyến tính ở khoảng liều gây độc.
Những đặc điểm của dược động học không tuyến tính bao gồm:
Nồng độ giảm theo thời gian sau khi tiêm một liều bolus của thuốc này không tuân
theo hàm số mũ. Thay vào đó, quá trình đào thải ban đầu diễn ra chậm và tăng lên
khi nồng độ trong huyết tương giảm (Hình 5.6).
Thời gian để đào thải 50% liều thuốc tăng khi tăng liều, do đó, thời gian bán thải
không phải không đổi đối với một thuốc mà phụ thuộc vào liều dùng ban đầu của thuốc.
Tăng liều thuốc không tăng tỷ lệ với lượng thuốc trong cơ thể một khi quá trình
đào thải thuốc được bảo hòa. Điều này cực kì quan trọng trên lâm sàng khi dùng
nồng độ huyết tương (ví dụ với thuốc phenytoin) để điều chỉnh liều thuốc. 45
Hình 5.6. Dược động học không tuyến tính: đường cong nồng độ thuốc trong
huyết tương-thời gian sau khi tiêm bolus một liều thuốc được đào thải
theo phương trình Michaelis-Menten
Câu hỏi lượng giá:
1. Phân tích sự khác nhau giữa mô hình dược động học một ngăn có quá trình thải trừ
bậc 1 và mô hình dược động học hai ngăn có quá trình thải trừ không tuyến tính.
2. Hiểu và ứng dụng tính toán các thông số dược động học cơ bản (như sinh khả dụng,
thể tích phân bố, độ thanh thải, thời gian bán thải), khoảng cách đưa thuốc, liều tấn
công, liều duy trì đối với 3 cách đưa thuốc: truyền tĩnh mạch với tốc độ không đổi,
tiêm bolus liều đơn duy nhất, tiêm bolus liều lặp lại.
3. Một bệnh nhân mắc bệnh suy tim được điều trị bằng digoxin và sau đó phải thêm
furosemid do suy tim sung huyết. Độ thanh thải là 1l/phút. Trước khi dùng furosemid,
Vd là 130l, sau khi dùng furosemid, Vd giảm còn 87l. Hãy cho biết thời gian bán thải
t1/2 và thời gian thuốc đạt Css thay đổi như thế nào trong thời gian trước và sau khi bệnh nhân dùng furosemid?
4. Một bệnh nhân nữ 55 tuổi được truyền 80mg gentamicin, cách 8 giờ một lần. Sau 30
phút ngừng đưa thuốc, lấy mẫu máu xác định nồng độ thuốc là 5,2mcg/ml. 7h sau
ngừng truyền, đo được nồng độ thuốc là 1,7mcg/ml. Tính các giá trị sau: a. t1/2
b. Nồng độ thuốc ở giờ thứ 8 sau khi ngừng đưa thuốc
c. Tính thời gian cần thiết nồng độ thuốc giảm từ 5,5mcg/ml tới 0,5mcg/ml 46
5. Một bệnh nhân nhi nam bị động kinh tự phát và điều trị bằng phenytoin, 200mg hàng
ngày, dùng liều đơn buổi tối. Sau một tuần, nồng độ phenytoin trong huyết thanh của
bệnh nhân là 25µmol/l. Khoảng liều điều trị trong huyết thanh là 40-80µmol/l. Liều
được tăng lên 300mg/ngày. Một tuần sau, bệnh nhân phàn nàn bị loạng choạng,
chứng giật cầu mắt, nồng độ phenytoin trong huyết tương là 125µmol/l. Liều được
giảm xuống 250mg/ngày. Triệu chứng tiến triển từ từ và nồng độ phenytoin trong
huyết tương giảm xuống 60µmol/l (trong khoảng điều trị). Giải thích mối liên hệ giữa
liều dùng, nồng độ thuốc trong huyết tương và triệu chứng lâm sàng trên bệnh nhân này? 47
CHƯƠNG 2. SỰ THAY ĐỔI CÁC THÔNG SỐ DƯỢC ĐỘNG HỌC Ở BỆNH
NHÂN SUY GIẢM CHỨC NĂNG GAN, THẬN - ỨNG DỤNG ĐIỀU CHỈNH LIỀU
THUỐC TRÊN LÂM SÀNG
BÀI 6: SỰ THAY ĐỔI CÁC THÔNG SỐ DƯỢC ĐỘNG HỌC Ở BỆNH NHÂN
SUY GAN-ỨNG DỤNG ĐIỀU CHỈNH LIỀU THUỐC TRÊN LÂM SÀNG MỤC TIÊU
1. Trình bày được sự thay đổi về các thông số dược động học ở bệnh nhân suy giảm chức năng gan.
2. Ứng dụng trong điều chỉnh liều thuốc ở bệnh nhân suy giảm chức năng gan. NỘI DUNG
1. Bệnh gan và sự suy giảm chức năng gan
Bệnh viêm gan cấp tính là tình trạng viêm của gan gây ra bởi virus hoặc các độc tố
trong gan. Trong viêm gan virus cấp tính, mức độ viêm thường nhẹ và thoáng qua, mặc
dù có thể chuyển thành thể mạn tính với biến chứng nghiêm trọng dẫn đến xơ gan hoặc tử
vong. Ở bệnh nhân viêm gan cấp tính, quá trình chuyển hóa và thải trừ thuốc bình thường
hoặc ít bị suy giảm, phụ thuộc vào mức độ tổn thương của tế bào gan. Nếu bệnh được
điều trị khỏi, các quá trình trên sẽ trở lại bình thường. Bệnh gan mạn tính thường là thứ
phát do nghiện rượu mạn tính hoặc viêm gan virus mạn tính, tế bào gan bị tổn thương
nghiêm trọng gây ảnh hưởng lớn đến quá trình chuyển hóa và thải trừ thuốc.
Xơ gan xảy ra thường xuyên nhất trong trường hợp mắc bệnh gan do rượu và là
biến chứng cuối cùng của các bệnh gan mạn tính. Bệnh xơ gan được đặc trưng bởi sự xuất
hiện của các sợi nguyên bào và lắng đọng collagen. Hậu quả làm giảm kích thước gan và
do đó làm giảm lượng enzyme cytochrome P450 trong gan. Sự lắng đọng của các mảng
xơ phá vỡ cấu trúc mạch máu gan bình thường và làm tăng tính kháng thành mạch và áp
lực tĩnh mạch cửa. Nguyên nhân này có thể làm giảm lưu lượng máu qua tĩnh mạch cửa,
thông thường chiếm 70% lưu lượng máu qua gan.
2. Vai trò của gan trong chuyển hóa và thải trừ thuốc
Hầu hết các thuốc tan trong lipid được chuyển hóa ít nhiều ở gan. Các phản ứng
chuyển hóa Giai đoạn I (Phase I) như các phản ứng oxy hóa, phản ứng thủy phân và phản
ứng khử thường qua trung gian là hệ thống enzyme cytocrom P-450 (CYP) được liên kết
với màng của mạng lưới nội chất bên trong tế bào gan. Các phản ứng Giai đoạn II (Phase
II) bao gồm các phản ứng liên hợp glucuronide, acetate và sulfate, một phần được xúc tác
qua trung gian là các enzyme của tế bào gan. Các phản ứng chuyển hóa thuốc Giai đoạn I 48
và Giai đoạn II thường tạo thành các chất chuyển hóa thân nước hơn và dễ được loại trừ
qua thận. Các protein vận chuyển như P- glycoprotein, chủ động bài tiết phân tử thuốc vào trong mật.
Gan nhận được nguồn cung cấp máu thông qua động mạch gan, chứa máu mang
oxy từ động mạch chủ qua động mạch mạc treo trên và tĩnh mạch cửa (Hình 6.1). Trung
bình lưu lượng máu qua gan 1-1,5 lít/phút ở người lớn với khoảng một phần ba đến từ
động mạch gan và khoảng hai phần ba đến từ các tĩnh mạch cửa. Thuốc đường uống phải
đi qua gan trước khi vào hệ tuần hoàn, vì vậy nếu thuốc được chuyển hóa ở gan, một phần
có thể bị mất hoạt tính bởi hiệu ứng chuyển hóa qua gan lần đầu (First- pass effect) trước
khi thể hiện tác dụng dược lý. Ngoài chuyển hóa ở gan, thuốc có thể được thải trừ dưới
dạng không thay đổi từ gan qua đường mật. Độ thanh thải của thuốc qua gan được tính theo phương trình sau: (1) Trong đó:
Cl : Độ thanh thải của thuốc qua gan H
LBF: Lưu lượng máu qua gan
fB: Tỷ lệ của thuốc tự do trong máu Cl' Độ thanh thải nội tại int: EH: Hệ số trích ly
Chuyển hóa thuốc qua gan chưa phát triển hoàn thiện ở trẻ sơ sinh đủ tháng (~ 40
tuần tuổi thai) và tiếp tục hoàn thiện đến mức ổn định khi trẻ được 3-6 tháng tuổi. Ở trẻ
sinh thiếu tháng (< 35 tuần ), chức năng chuyển hóa thuốc ở gan cần nhiều thời gian hơn
để được hoàn thiện. Quá trình chuyển hóa thuốc trên cơ thể trẻ em thường nhanh hơn trên
cơ thể trẻ tuổi dậy thì, tính trên cơ sở trọng lượng cơ thể. Tốc độ chuyển hóa chậm dần
cho tới tuổi trưởng thành. Ở bệnh nhân trên 65 tuổi, độ thanh thải qua gan của một số
thuốc có thể giảm, tuy nhiên do tình trạng bệnh làm thay đổi tác dụng dược lý của thuốc
nên sự ảnh hưởng do tuổi cao không rõ ràng. Ngoài ra, ở đối tượng này, khối lượng gan
giảm, chức năng chuyển hóa thuốc của các tế bào gan còn lại cũng giảm. 49
Hình 6.1. Sơ đồ dòng máu qua gan
Ở bệnh nhân viêm gan, các tế bào gan có thể bị giảm khả năng hoạt động hoặc bị
chết. Ở bệnh nhân viêm gan cấp tính, chuyển hóa thuốc thường bị giảm nhẹ, tuy nhiên
không đòi hỏi phải giảm liều thuốc. Nếu viêm gan cấp tính tiến triển thành viêm gan mạn
tính, tế bào gan bị tổn thương không hồi phục, khi đó bắt buộc phải có sự điều chỉnh liều lượng thuốc.
Ở bệnh nhân xơ gan, các tế bào gan suy giảm chức năng, đòi hỏi có sự điều chỉnh
liều lượng thuốc cho bệnh nhân, đặc biệt với bệnh nhân xơ gan nặng. Nếu quá trình tổn
thương của các tế bào gan kéo dài, bệnh viêm gan có thể tiến triển thành bệnh xơ gan. Khi
tế bào gan bị tổn thương không còn khả năng chuyển hóa thuốc hiệu quả, độ thanh thải
của thuốc qua gan sẽ giảm. Nếu thuốc phải trải qua hiệu ứng chuyển hóa qua gan lần đầu,
lượng thuốc được chuyển hóa giảm xuống, lượng thuốc chưa chuyển hóa tăng lên, dẫn
đến tăng sinh khả dụng của thuốc.
Sự suy giảm đồng thời độ thanh thải qua gan và lượng thuốc được chuyển hóa qua
gan lần đầu sẽ làm tăng nồng độ thuốc ở trạng thái ổn định trong máu đối với các thuốc
dùng đường uống. Ở bệnh nhân suy gan, lưu lượng máu qua gan giảm do các tế bào gan
bị thay thế bằng các mô liên kết không có chức năng, gây tăng áp lực tĩnh mạch cửa và và
hạn chế dòng máu tới gan. Dòng máu qua gan giảm dẫn tới giảm lượng thuốc tiếp xúc với
các tế bào gan còn hoạt động và làm giảm độ thanh thải của thuốc qua gan. Gan là cơ
quan sản xuất albumin và α1-acid glycoprotein, hai protein chính liên kết các thuốc có
tính acid và base tương ứng trong máu. Ở bệnh nhân bị xơ gan, việc sản xuất các protein
này bị suy giảm. Khi đó, lượng thuốc tự do không liên kết với protein trong máu tăng lên.
Ngoài ra, các chất nội sinh có trong máu cũng thường được đào thải qua gan ví dụ như
bilirubin, chúng có thể cạnh tranh liên kết protein huyết tương với các thuốc và đẩy các
phân tử thuốc ra khỏi vị trí liên kết. Do đó, tỷ lệ thuốc tự do trong máu tăng lên và làm
thay đổi độ thanh thải thuốc của gan và thận, đồng thời thay đổi thể tích phân bố các loại
thuốc có ái lực cao với protein. Mối liên hệ được miêu tả bằng phương trình sau đây: 50 V = VB + (fB / fT) VT (2) Trong đó: V: Thể tích phân bố,
VB và VT: Thể tích sinh lý của máu và các mô tương ứng f : Tỷ lệ B và fT
thuốc dạng tự do trong máu và các mô tương ứng
Do độ thanh thải thuốc giảm và thể tích phân bố trong cơ thể thường tăng hoặc
thay đổi không đáng kể ở bệnh nhân mắc bệnh gan, hằng số tốc độ thải trừ luôn giảm đi ở
những bệnh nhân này (ke = Cl/V, trong đó Cl là độ thanh thải và V là thể tích phân bố).
Xác định thang điểm Child-Pugh (Child-Pugh score)
Khác với trường hợp của bệnh suy thận, không có một thông số lâm sàng duy nhất
nào có thể giúp đánh giá chức năng gan giống như độ thanh thải creatinin trong trường
hợp đánh giá chức năng thận. Cách phổ biến nhất để đánh giá khả năng chuyển hóa thuốc
của gan là xác định điểm số Child-Pugh cho một bệnh nhân. Điểm số Child-Pugh dựa trên
năm thông số hóa sinh và triệu chứng lâm sàng. Năm thông số này là albumin huyết
thanh, bilirubin toàn phần, thời gian prothrombin kéo dài, bệnh cổ trướng và bệnh não
gan. Mỗi thông số này được cho điểm từ 1 (bình thường) đến 3 (bất thường nghiêm trọng)
(Bảng 4.1), điểm số Child-Pugh là tổng điểm của 5 thông số trên. Điểm số Child-Pugh
cho một bệnh nhân có chức năng gan bình thường là 5 trong khi số điểm cho một bệnh
nhân với các giá trị bất thường của albumin huyết thanh, bilirubin toàn phần và thời gian
prothrombin có kèm theo bệnh cổ trướng nặng và bệnh não gan là 15.
Bảng 6.1. Thang điểm Child-Pugh cho bệnh nhân mắc bệnh gan
Thông số/triệu chứng 1 điểm 2 điểm 3 điểm
Bilirubin toàn phần (mg/100ml) <2,0 2,0-3,0 >3,0
Albumin huyết thanh (mg/100ml) >3,5 2,8-3,5 <2,8
Thời gian prothrombin (giây) <4 4-6 >6 Cổ trướng Không Nhẹ Trung bình Bệnh não gan Không Trung bình Nặng
Điểm số Child-Pugh bằng 8-9 là căn cứ để giảm nhẹ (25%) liều thuốc khởi đầu
hàng ngày đối với các thuốc chủ yếu được chuyển hóa qua gan (≥ 60%). Điểm số Child- 51
Pugh ≥ 10 là căn cứ giảm 50% liều thuốc khởi đầu hàng ngày đối với các thuốc chủ yếu chuyển hóa qua gan.
Đối với bất kỳ bệnh nhân có hoặc không có rối loạn chức năng gan, liều khởi đầu
có nghĩa là liều xuất phát để điều chỉnh liều cho những lần dùng thuốc sau dựa trên cơ sở
đáp ứng của bệnh nhân và các tác dụng phụ của thuốc. Ví dụ, liều thông thường của một
loại thuốc, được chuyển hóa qua gan 95%, là 500 mg/6 giờ và tổng liều hàng ngày là
2000 mg/ngày . Đối với một bệnh nhân xơ gan có điểm số Child-Pugh 12, liều khởi đầu
phù hợp sẽ là 50% liều thông thường, có nghĩa là 1000 mg/ngày. Bệnh nhân sẽ dùng
thuốc theo 2 cách: 250 mg/6 giờ hoặc 500 mg/12 giờ. Bệnh nhân sẽ được theo dõi chặt
chẽ các tác dụng dược lý và độc tính do thuốc, liều lượng thuốc có thể được điều chỉnh lại khi cần thiết.
3. Nồng độ thuốc trong huyết tương và tác dụng của thuốc
Ở bệnh nhân có rối loạn chức năng gan, các thông số dược động học thay đổi dẫn
đến sự thay đổi phức tạp về nồng độ thuốc toàn phần và nồng độ thuốc dạng tự do ở trạng
thái ổn định và tác dụng của thuốc. Những thay đổi này phụ thuộc vào việc thuốc có tỷ lệ
trích ly qua gan cao hay thấp (Phương trình 1).
Đối với thuốc có tỷ lệ trích ly qua gan thấp (≤ 30%), giá trị về số của lưu lượng
máu qua gan lớn hơn nhiều so với tích giữa phần thuốc không liên kết trong máu và độ
thanh thải nội tại của thuốc (LBF >> fB*Cl'int) và tổng mẫu số của phương trình trên xấp
xỉ lưu lượng máu qua gan [LBF ≈ LBF + (f
)]. Khi đó phương trình 1 tính độ thanh B*Cl'int
thải của thuốc qua gan có dạng như sau: (3)
Tương tự như vậy, đối với các thuốc có tỷ lệ trích ly qua gan cao (≥ 70%), giá trị về số
của lưu lượng máu qua gan nhỏ hơn nhiều so với tích của phần thuốc không liên kết trong
máu với độ thanh thải nội tại của thuốc (LBF << fB*Cl'int) và tổng mẫu số của phương
trình xấp xỉ bằng tích của phần thuốc không liên kết với độ thanh thải nội tại của thuốc [fB
⋅ Cl 'int ≈ LBF + (fB*Cl'int)]. Khi đó phương trình 1 tính độ thanh thải của thuốc qua gan có dạng như sau: (4) 52
Đối với thuốc có tỷ lệ trích ly qua gan trung bình (30%-70%), phương trình tính độ
thanh thải của gan liên quan đến tất cả ba yếu tố: lưu lượng máu qua gan, phần thuốc dạng
tự do trong máu và độ thanh thải nội tại của thuốc. Hình 6.2 minh họa sự thay đổi của
nồng độ thuốc ổn định trong máu (Css) và tác dụng dược lý (Effect) trên bệnh nhân bị
bệnh gan. Sự gia tăng của các các thông số được ký hiệu bằng đường gấp khúc đi lên và
sự giảm của các thông số là đường gấp khúc đi xuống. Độ thanh thải nội tại (Cl’int) suy
giảm do các tế bào gan bị hủy hoại, lưu lượng dòng máu qua gan (LBF) và phần thuốc
không liên kết trong máu (fB) không bị thay đổi (Hình 6.2). Sự thay đổi này sẽ làm giảm
độ thanh thải của gan, thể tích phân bố (V) không thay đổi do thể tích máu và khối lượng
mô hoặc protein huyết tương và liên kết mô không thay đổi. Do vậy, thời gian bán thải
tăng lên do độ thanh thải giảm [t1/2= ( 0,693*Vd)/Cl]. Nồng độ thuốc ổn định trong máu
dạng toàn phần và dạng tự do sẽ tăng song song, đáp ứng dược lý sẽ tăng do nồng độ
thuốc tự do trong máu tăng.
Hình 6.2. Sự thay đổi các thông số động học đối với các thuốc có tỷ lệ trích
ly qua gan trung bình khi độ thanh thải nội tại thay đổi.
Trong trường hợp thuốc giảm liên kết với protein huyết tương do giảm lượng
albumin, lưu lượng máu qua gan và độ thanh thải nội tại không thay đổi, tuy nhiên, phần 53
thuốc không liên kết trong máu sẽ tăng lên. Do phần thuốc không liên kết trong máu tăng
lên, độ thanh thải của gan và thể tích phân bố sẽ đồng thời tăng.
Trong trường hợp của các thuốc có hệ số trích ly qua gan thấp, độ thanh thải tăng
do tăng lượng thuốc tự do rời khỏi máu đi vào tế bào gan để được chuyển hóa. Thể tích
phân bố tăng lên do có nhiều hơn các phân tử thuốc tự do từ hệ tuần hoàn phân bố vào mô
cơ thể. Tùy thuộc vào những thay đổi tương đối của độ thanh thải và thể tích phân bố,
thời gian bán thải của thuốc có thể tăng, giảm hoặc không thay đổi. Nồng độ thuốc toàn
phần trong máu ở trạng thái ổn định sẽ giảm do độ thanh thải toàn phần tăng lên, tuy
nhiên nồng độ thuốc tự do sẽ không thay đổi do sự suy giảm nồng độ toàn phần bắt nguồn
từ sự tăng lượng thuốc tự do. Cuối cùng, tác dụng dược lý của thuốc không thay đổi do
nồng độ thuốc tự do ở trạng thái ổn định không thay đổi. Liên kết với protein suy giảm do
nồng độ thuốc toàn phần trong máu ở trạng thái ổn định giảm. Bác sĩ điều trị phải chú ý
tới trường hợp nồng độ thuốc toàn phần trong máu giảm nhưng nồng độ thuốc tự do
không thay đổi, tránh trường hợp tăng liều thuốc bất cẩn gây tác hại cho bệnh nhân. Đối
với một số thuốc liên kết mạnh với protein huyết tương như phenytoin, acid valproic và
carbamazepine, nồng độ thuốc tự do trong máu là cơ sở hướng dẫn việc điều chỉnh liều
thuốc ở bệnh nhân mắc bệnh gan.
Trong trường hợp các thuốc có tỷ lệ trích ly qua gan cao, giảm liên kết với protein
huyết tương khiến cho nồng độ thuốc tự do trong máu tăng cao, nhưng không có sự thay
đổi về lưu lượng máu qua gan hoặc độ thanh thải nội tại. Lưu lượng máu qua gan không
thay đổi dẫn đến độ thanh thải cũng không thay đổi. Phần thuốc tự do trong máu tăng lên
làm tăng thể tích phân bố, khiến cho thời gian bán thải tăng lên. Nồng độ thuốc toàn phần
ở trạng thái ổn định không thay đổi do độ thanh thải không thay đổi. Lượng thuốc tự do
trong máu ở trạng thái ổn định tăng lên do lượng thuốc tự do trong máu tăng, dẫn đến tác
dụng dược lý của thuốc tăng lên. Đây là sự thay đổi rất tinh tế trong chuyển hóa thuốc,
nồng độ thuốc toàn phần không thay đổi nhưng tác dụng dược lý của thuốc lại tăng lên.
Các bác sĩ phải lưu ý điều này. Nếu không có thông tin về nồng độ thuốc tự do trong máu
(hoặc không có thông tin về nồng độ thuốc toàn phần), cần thử nghiệm cho bệnh nhân bắt
đầu với liều thuốc thấp. Đối với thuốc đường uống, do nồng độ thuốc tự do trong máu
tăng, bác sĩ có thể lưu ý giảm liều cho bệnh nhân.
Nếu độ thanh thải nội tại giảm do tế bào gan bị phá hủy, lưu lượng dòng máu qua
gan và phần thuốc tự do trong máu không bị ảnh hưởng. Các hằng số dược động học cũng
không thay đổi do không bị ảnh hưởng bởi độ thanh thải nội tại. Do đó, nồng độ thuốc tự
do ổn định, nồng độ thuốc toàn phần trong máu và tác dụng dược lý không thay đổi. Đối
với các thuốc được dùng đường uống, chuyển hóa qua gan lần đầu giảm làm tăng sinh khả
dụng của thuốc. Do vậy, nồng độ thuốc toàn phần và thuốc tự do tăng lên, tác dụng dược 54
lý cũng tăng lên (Css=[F*(D/τ)/Cl], trong đó F là sinh khả dụng, Css là tổng nồng độ
thuốc ổn định, D là liều thuốc, τ là khoảng cách liều và Cl là độ thanh thải).
Giảm lưu lượng máu qua gan cần được lưu ý đối với các thuốc có tỷ lệ trích ly qua
gan thấp. Mặc dù lưu lượng máu qua gan giảm không làm thay đổi độ thanh thải nội tại,
liên kết với protein huyết tương, độ thanh thải hoặc thể tích phân bố, do đó, sẽ không làm
thay đổi nồng độ thuốc toàn phần trạng thái ổn định, nồng độ thuốc dạng tự do ổn định
hoặc tác dụng dược lý của thuốc. Tuy nhiên, sự sụt giảm mạnh lưu lượng máu qua gan sẽ
hạn chế đưa thuốc vào gan và làm thay đổi độ thanh thải của gan đối với nhiều thuốc có tỷ lệ trích ly thấp.
Những thay đổi về dược động học và dược lý rõ ràng hơn đối với các loại thuốc có
tỷ lệ trích ly qua gan cao (Hình 6.3). Lưu lượng máu qua gan giảm không làm thay đổi độ
thanh thải nội tại hoặc phần thuốc tự do trong máu. Đối với các thuốc này, độ thanh thải
giảm do phụ thuộc vào lưu lượng máu qua gan. Thể tích phân bố không thay đổi, trong
khi thời gian bán thải tăng do độ thanh thải giảm. Độ thanh thải giảm dẫn đến tăng nồng
độ thuốc toàn phần trong máu và do đó tăng nồng độ thuốc tự do trong máu, dẫn đến tăng
tác dụng dược lý của thuốc. Nếu thuốc dùng đường uống, chuyển hóa qua gan lần đầu
tăng làm giảm sinh khả dụng của thuốc, bù lại một phần cho sự tăng của nồng độ thuốc
toàn phần và thuốc tự do trong máu ở trạng thái ổn định.
Hình 6.3. Sự thay đổi các thông số độ 55
ng học đối với các thuốc có tỷ lệ trích
ly qua gan cao khi lưu lượng máu qua gan thay đổi.
4. Hiệu chỉnh liều lượng các thuốc chuyển hóa qua gan
Đối với các thuốc chủ yếu được chuyển hóa qua gan, các thông số dược động học
gắn liền với bệnh nhân mắc bệnh gan được tính toán bằng cách sử dụng các giá trị của các
thông số đã đo được trước đó trên bệnh nhân mắc cùng một loại bệnh gan (ví dụ: viêm
gan hoặc xơ gan) và có mức độ rối loạn chức năng gan tương tự. Bảng 6.2 cho biết giá trị
của độ thanh thải theophyllin trên nhiều bệnh nhân khác nhau bao gồm cả bệnh nhân mắc
bệnh xơ gan. Liều dùng thuốc và khoảng cách thời gian đưa liều nhằm đạt nồng độ thuốc
trong máu ổn định gần giới hạn dưới của cửa sổ điều trị, được tính toán bằng các phương
trình dược động học dựa trên các thông số dược động học. Ví dụ, tỉ lệ liều lượng
theophyllin được liệt kê trong Bảng 6.2 cho biết nồng độ theophyllin được tính toán để
đạt được nồng độ thuốc trong máu ổn định từ 8 đến 12 mg/l, được tính toán bằng cách
nhân độ thanh thải theophyllin với nồng độ thuốc trong máu ổn định mong muốn (MD =
Css x Cl, trong đó MD là liều duy trì, Css là nồng độ thuốc trong máu ổn định và Cl là độ
thanh thải của theophyllin). Độ thanh thải theophyllin trung bình thường chỉ khoảng 50%
ở người lớn mắc bệnh xơ gan so với người lớn có chức năng gan bình thường. Do vậy,
liều theophyllin khởi đầu cho bệnh nhân xơ gan thường là một nửa liều thông thường cho
bệnh nhân người lớn có chức năng gan bình thường.
Bảng 6.2. Điều chỉnh liều theophyllin theo độ thanh thải ở các điều kiện khác nhau
Tình trạng bệnh/điều kiện
Độ thanh thải trung bình Liều trung bình (ml/phút/kg) (mg/kg/h) Trẻ em 1-9 tuổi 1,4 0,8
Trẻ em 9-12 tuổi hoặc người 1,25 0,7
trưởng thành có hút thuốc lá
Thiếu niên 12-16 tuổi hoặc người 0,9 0,5
già (>65 tuổi) có hút thuốc lá
Người trưởng thành không hút 0,7 0,4 thuốc lá
Người già không hút thuốc lá 0,5 0,3
Bệnh nhân suy tim mất bù, tắc 0,35 0,2
nghẽn phổi mạn tính, xơ gan
Khi kê đơn cho bệnh nhân mắc bệnh gan các thuốc được đào thải chủ yếu qua gan,
có thể giảm liều thuốc trong khi vẫn giữ khoảng cách liều bình thường hoặc duy trì liều
bình thường và kéo dài khoảng cách đưa liều hoặc thay đổi cả liều và khoảng cách đưa 56
liều. So với người có chức năng gan bình thường, ở người bệnh có suy giảm chức năng
gan, chỉ định một liều bình thường với khoảng cách đưa liều dài hơn sẽ tạo ra nồng độ
thuốc ổn định trong máu tối thiểu và tối đa tương tự. Tuy nhiên, nếu giảm liều dùng và
giữ nguyên khoảng cách đưa liều, nồng độ thuốc ổn định trong máu tối đa sẽ thấp hơn và
nồng độ thuốc ổn định tối thiểu sẽ cao hơn ở bệnh nhân mắc bệnh gan so với bệnh nhân
có chức năng gan bình thường.
5. Một số nguyên tắc khi dùng thuốc cho người bệnh suy gan
Giảm lượng thuốc cần dùng ở mức tối thiểu.
Tránh thuốc gây độc cho gan.
Điều chỉnh liều dùng của nhiều loại thuốc cho người bệnh suy chức năng gan
để tránh ngộ độc cho gan.
Gan phản ứng bù trừ bằng cách to ra, bệnh gan trở nên trầm trọng trước khi
thấy những thay đổi quan trọng trong chuyển hoá thuốc. Các xét nghiệm chức
năng gan thường quy ít có tác dụng trong việc đánh giá khả năng chuyển hoá thuốc của gan.
Ở trẻ sơ sinh thiếu tháng, chức năng gan chưa phát triển đầy đủ, do đó phải
thận trọng khi dùng thuốc ở những đối tượng này.
Những thuốc cần tránh hoặc thận trọng khi sử dụng
Thuốc chống hen: aminophylin, theophylin
Thuốc chữa tiểu đường: glibenclamid, gliclazid, metformin
Thuốc chống nấm: ketoconazol, griseofulvin
Thuốc kháng histamin: clorpheniramin, promethazin, diphenhydramin, dimenhydrinat.
Thuốc chống ung thư: cyclophosphamid, cytarabin, doxorubicin, methotrexat, vinblastin, vincristin.
Thuốc chống lao: isoniazid, pyrazinamide, rifampicin
Thuốc ngủ: diazepam
Kháng sinh nhóm bêta-lactam: ceftriaxon, cloxacilin
Thuốc lợi niệu nhóm thiazide và thuốc lợi niệu quai henle: furosemid, hydroclorothiazid.
Kháng sinh nhóm macrolid: erythromycin, clarithromycin, azithromycin 57
Thuốc chống viêm không steroid (NSAID): acetylsalicylic acid, diclofenac,
ibuprofen, indomethacin, ketoprofen, meloxicam, naproxen, piroxicam, tenoxicam.
Thuốc giảm đau nhóm opiat: morphin, pethidin, fentanyl, dextropropoxyphen, codein, dextromethorphan.
Thuốc chống đông máu đường uống: warfarin
Thuốc tránh thai đường uống Paracetamol
Nhóm quinolon: ciprofloxacin, nalidixic acid, norfloxacin, ofloxacin
Các kháng sinh khác: tetracyclin, cloramphenicol, metronidazol, clindamycin.
Thuốc chống tăng mỡ máu nhóm statin: simvastatin Câu hỏi lượng giá
Một bệnh nhân nam, 62 tuổi, cân nặng 65 kg mắc bệnh xơ gan có nồng độ bilirubin
toàn phần 2,6 mg/dl, albumin huyết thanh 2,5 mg/dl, thời gian prothrombin kéo dài hơn
bình thường 8 giây, có triệu chứng bụng trướng nhẹ và không mắc bệnh não gan. Bệnh
nhân bị tắc nghẽn phổi mãn tính nghiêm trọng cần được chỉ định theophyllin. Biết bệnh
nhân này không hút thuốc lá và không có suy tim. Tính điểm số ChildPugh của bệnh
nhân, độ thanh thải của theophyllin và liều theophyllin để đạt được nồng độ thuốc trong
máu ở trạng thái ổn định là 10 mg/l. 58
BÀI 7: SỰ THAY ĐỔI CÁC THÔNG SỐ DƯỢC ĐỘNG HỌC Ở BỆNH NHÂN
SUY THẬN-ỨNG DỤNG ĐIỀU CHỈNH LIỀU THUỐC TRÊN LÂM SÀNG MỤC TIÊU
1. Trình bày được sự thay đổi về các thông số dược động học ở bệnh nhân suy giảm chức năng thận.
2. Ứng dụng trong điều chỉnh liều thuốc ở bệnh nhân suy giảm chức năng thận. NỘI DUNG 1. Bệnh suy thận
Bệnh suy thận được đặc trưng bởi sự tổn thương cấu trúc của thận hoặc sự bất bình
thường của thành phần nước tiểu, dẫn đến suy giảm chức năng lọc của cầu thận. Bệnh suy
thận được định nghĩa khi mức độ lọc cầu thận giảm (Glomerular Filtration Rate: GFR) <
60ml/phút/1,73m2. Suy thận được chia thành 5 cấp độ:
Suy thận độ 1 (GFR>=90ml/phút/1,73m2 có kèm theo bằng chứng tổn
thương cấu trúc thận hoặc bất thường trong thành phần nước tiểu).
Suy thận độ 2 (GFR: 60-89ml/phút/1,73m2 có kèm theo bằng chứng tổn
thương cấu trúc thận hoặc bất thường trong thành phần nước tiểu).
Suy thận độ 3 (GFR: 30-59ml/phút/1,73m2 có hoặc không kèm theo bằng
chứng tổn thương cấu trúc thận hoặc bất thường trong thành phần nước tiểu).
Suy thận độ 4 (GFR: 15-29ml/phút/1,73m2 có hoặc không kèm theo bằng
chứng tổn thương cấu trúc thận hoặc bất thường trong thành phần nước tiểu).
Suy thận độ 5 (GFR<15ml/phút/1,73m2).
Sự suy giảm chức năng thận dẫn đến khả năng lọc sạch thuốc khỏi huyết tương của
thận giảm, thuốc dễ tích lũy trong cơ thể, có thể gây độc tính tích lũy.
Sự biến đổi các thông số dược động học liên quan đến sự suy giảm chức năng thận:
Tổn thương thận làm tuần hoàn máu bị ứ trệ và cơ thể bị phù dẫn đến sinh khả
dụng của các thuốc theo đường tiêm bắp và tiêm dưới da bị ảnh hưởng. Với thuốc đưa
theo đường uống, tổn thương chức năng thận làm tăng sinh khả dụng của những thuốc có
hệ số chiết xuất cao, chịu sự khử hoạt mạnh ở vòng tuần hoàn đầu (propranolol,
verapamil, các hormon) do bão hòa khả năng phá hủy thuốc ở gan và do nồng độ thuốc
trong máu tăng cao và do ứ trệ tuần hoàn. 59
Một số tổn thương bệnh lý ở thận gây giảm lượng albumin trong huyết thanh, thay
đổi cấu trúc một số protein của huyết tương. Suy thận cũng gây ứ trệ các chất nội sinh
(ure, creatinin, các acid béo…) cạnh tranh với thuốc trong liên kết với protein huyết
tương, làm tăng nồng độ thuốc ở dạng tự do. Ngoài ra, cùng với sự tăng thể tích chất lỏng
ngoại bào dẫn đến tăng thể tích phân bố của nhiều thuốc.Tuy nhiên, trong một số trường
hợp suy thận thể tích phân bố của thuốc lại giảm như sử dụng thuốc lợi tiểu quá liều, suy tuyến thượng thận.
Sự ứ trệ các chất chuyển hóa khi suy thận có thể làm tăng bài xuất qua mật các
dạng liên hợp với acid glucuronic của những thuốc được bài xuất nhiều ở dạng này
(oxazepam, diflunisal…). Quá trình này ảnh hưởng đến độ thanh thải thuốc qua thận mà hậu quả là giảm ClR.
Các thuốc bài xuất qua thận > 50% ở dạng còn hoạt tính có t1/2 tăng rõ rệt khi sức
lọc cầu thận <30ml/phút. Đối với những thuốc bị chuyển hóa gần như 100% ở gan thì t1/2
không thay đổi khi thiểu năng thận.
2. Vai trò của thận trong chuyển hóa và thải trừ thuốc
Thận là cơ quan thải trừ thuốc chính của cơ thể. Quá trình thải trừ thuốc và tái hấp
thu thuốc tại ống thận giảm khi GFR giảm do sự tổn thương của các cầu thận. Hầu hết các
thuốc dễ tan trong nước được đào thải qua thận dưới dạng không biến đổi ở một mức độ
nào đó. Với các thuốc khó tan trong nước, quá trình chuyển hóa biến thuốc thành các chất
dễ tan trong nước hơn nhờ các quá trình oxy hóa hoặc liên hợp, sau đó dễ dàng được đào
thải qua thận. Cầu thận là đơn vị chức năng của thận có chức năng đào thải các chất thải
đồng thời đào thải các phân tử thuốc ra khỏi cơ thể. Các phân tử thuốc không liên kết có
kích thước tương đối nhỏ và được lọc qua cầu thận. Quá trình lọc qua cầu thận là con
đường thải trừ chính của nhiều loại thuốc. Thuốc có thể được bài tiết chủ động vào nước
tiểu ở ống lượn gần. Quá trình bài tiết này nhờ vào các chất mang hoặc các bơm đặc thù,
vận chuyển thuốc từ các mạch máu gần với cầu thận vào phần ống lượn gần của thận.
Song song với quá trình đào thải, một số loại thuốc có thể được tái hấp thu từ nước tiểu
trở lại vào máu qua thận. Quá trình tái hấp thu là quá trình thụ động và đòi hỏi phân tử
thuốc có tính thân lipid nhất định. Vì vậy, tái hấp thu ở ống thận chịu ảnh hưởng của độ
pH của nước tiểu, pKa của phân tử thuốc và mức độ ion hóa của phân tử thuốc. Thường
những hợp chất không ion hóa trong nước tiểu là chất thân lipid, chúng có thể đi qua
màng lipid và dễ bị tái hấp thu ở ống thận. Phương trình sau đây mô tả quá trình thải trừ một chất qua thận: 60 Trong đó: fB:
Phần thuốc tự do trong máu
GFR: Mức độ lọc cầu thận
RBF: Lưu lượng máu qua thận
Cl'sec: Độ thanh thải nội tại của thuốc không liên kết được bài tiết qua ống thận FR:
Tỷ lệ thuốc được tái hấp thu
Ở trẻ sơ sinh đủ tháng (~ 40 tuần tuổi), chức năng thận chưa hoàn toàn phát triển.
Chức năng thận phát triển hoàn thiện ở độ tuổi từ 3-6 tháng sau khi sinh. Ở trẻ sinh thiếu
tháng (< 35 tuần), quá trình này có thể kéo dài lâu hơn. Ở người trưởng thành khỏe mạnh
18-22 tuổi, chức năng thận, được đo bằng mức độ lọc cầu thận, thường trung bình khoảng
120-140 ml/phút. Chức năng thận giảm dần theo tuổi và đạt mức trung bình 50-60
ml/phút ở độ tuổi 65 và 30-40 ml/phút ở độ tuổi 80. Đó là giới hạn bình thường thường
được dùng trong lâm sàng. Ở những bệnh nhân có bệnh thận, chức năng cầu thận suy giảm.
Tùy thuộc cơ chế bệnh sinh của bệnh thận, ở bệnh nhân suy thận cấp tính, chức
năng thận có thể phục hồi sau một thời gian điều trị hỗ trợ và lọc máu nhân tạo. Ở bệnh
nhân suy thận cấp do giảm đột ngột lưu lượng máu thận ví dụ hạ huyết áp, sốc hoặc thể
tích máu giảm hoặc do sử dụng các thuốc gây độc cho thận như thuốc kháng sinh
aminoglycosid hoặc vancomycin, chức năng thận chỉ được phục hồi nếu quá trình trị liệu
rối loạn chức năng thận thành công. Ở bệnh nhân suy thận mạn tính, chức năng cầu thận
suy giảm không hồi phục nên chức năng thận không phục hồi được trạng thái bình thường.
Bác sĩ cần xem xét việc thay đổi liều thuốc đang dùng cho bệnh nhân mắc bệnh
thận. Nói chung, bác sĩ có thể xem xét giảm nhẹ liều thuốc khi độ thanh thải creatinin
<50-60 ml/phút, giảm vừa phải khi độ thanh thải creatinin là <25-30 ml/phút và giảm
đáng kể liều thuốc khi độ thanh thải creatinin ≤ 15 ml/phút . Để thay đổi liều thuốc cho
bệnh nhân suy thận, có thể dùng phương pháp giảm liều thuốc và giữ lại khoảng cách liều
dùng như thông thường hoặc giữ liều thuốc như thông thường và tăng khoảng cách liều
thuốc hoặc đồng thời vừa giảm liều thuốc vừa tăng khoảng cách liều. Phương pháp sử
dụng phụ thuộc vào đường dùng thuốc, dạng bào chế có sẵn và đáp ứng dược lực học của
cơ thể đối với thuốc.Ví dụ, nếu thuốc được chỉ định dùng đường uống và chỉ có một
lượng hạn chế dạng bào chế rắn có sẵn, phương pháp được sử dụng là giữ liều thông
thường và tăng khoảng cách liều. Nếu thuốc được chỉ định dùng đường tiêm, phương
pháp được sử dụng là giảm liều và giữ nguyên khoảng cách liều dùng ban đầu. 61
Đối với các thuốc có cửa sổ điều trị hẹp như thuốc kháng sinh aminoglycosid và
vancomycin, những thuốc cần được thiết lập khoảng liều an toàn giữa nồng độ ổn định
(Css) nhỏ nhất và nồng độ ổn định cao nhất, liều lượng thuốc và khoảng cách đưa liều
cần được tính toán phù hợp để đạt được mục tiêu điều trị. Nếu giảm liều thuốc và giữ
nguyên khoảng cách liều, nồng độ thuốc tối đa thường thấp hơn và nồng độ thuốc tối
thiểu cao hơn ở những bệnh nhân suy giảm chức năng thận so với những bệnh nhân có
chức năng thận bình thường được chỉ định liều thuốc thông thường. Nếu tăng khoảng
cách liều và giữ nguyên liều lượng thuốc, nồng độ thuốc tối đa và tối thiểu ở bệnh nhân
suy thận và bệnh nhân có chức năng thận bình thường được chỉ định liều thường dùng là giống nhau.
Hình 7.1 miêu tả sự thay đổi của nồng độ thuốc trong huyết tương theo thời gian.
Đường nét liền là đồ thị của bệnh nhân có chức năng thận bình thường, liều dùng
300mg/6giờ. Đường nét đứt là đồ thị của bệnh nhân có rối loạn chức năng thận, liều dùng
300mg/12giờ, nghĩa là giữ nguyên liều và tăng gấp đôi khoảng cách đưa liều. Đường
chấm nhỏ là đồ thị của bệnh nhân có rối loạn chức năng thận, liều dùng 150mg/6giờ,
nghĩa là giảm 1/2 liều và giữ nguyên khoảng cách đưa liều. Đồ thị đường nét liền và
đường nét đứt gần tương tự nhau, có nghĩa ở bệnh nhân mắc bệnh thận, nếu giữ nguyên
liều và tăng khoảng cách đưa liều (300mg/12giờ), nồng độ đỉnh (peak) và nồng độ đáy
(trough) ở trạng thái ổn định (Css) ở người bình thường và người mắc bệnh thận là tương
tự nhau. Trong khí đó, nồng độ đỉnh lại thấp hơn và nồng độ đáy lại cao hơn ở người mắc
bệnh thận giảm liều và giữ nguyên khoảng cách đưa liều (150mg/6giờ) so với trường hợp
giữ liều và tăng khoảng cách đưa liều (300mg/12giờ).
Kể từ giữa những năm 1980, FDA đã yêu cầu phải thực hiện các nghiên cứu dược
động học đối với các thuốc được đào thải qua thận ở những bệnh nhân có độ thanh thải
creatinin giảm trước khi được FDA chấp thuận cho phép lưu hành trên thị trường. Trong
những trường hợp này, tờ hướng dẫn sử dụng thuốc có thể cung cấp hướng dẫn về liều khởi đầu hợp lý. 62
Hình 7.1. Sự thay đổi của nồng độ đỉnh (peak) và nồng độ đáy (trough) của
thuốc khi điều chỉnh liều ở bệnh nhân mắc bệnh thận
3. Hướng dẫn hiệu chỉnh liều thuốc ở bệnh nhân suy thận
Mức độ suy thận được đánh giá qua hệ số suy giảm chức năng thận RF Trong đó:
Clcr-st: Độ thanh thải creatinin ở bệnh nhân suy thận.
Clcr-bt: Độ thanh thải creatinin ở bệnh nhân có chức năng thận bình thường (80-120 ml/ph).
Để đánh giá mức độ suy thận, người ta tiến hành xét nghiệm mức creatinin trong huyết
tương và tính Clcr-st (ml/phút) từ công thức Cockcroft & Gault như sau:
Đánh giá mức độ giảm bài xuất thuốc ở người suy thận so với người bình thường Tính hệ số Q: 63 Trong đó:
Q: Hệ số hiệu chỉnh cho bệnh nhân có suy giảm chức năng thận.
fe: Tỷ lệ thuốc được bài xuất qua thận ở dạng còn hoạt tính.
RF: Hệ số suy giảm chức năng thận
Hiệu chỉnh liều ở bệnh nhân suy thận Có 3 cách hiệu chỉnh
3.1.Giữ nguyên khoảng cách đưa thuốc và giảm liều
3.2. Giữ nguyên liều nhưng nới rộng khoảng cách đưa thuốc
3.3. Vừa giảm liều, vừa nới rộng khoảng cách đưa thuốc
Nhiều trường hợp, nếu dùng hệ số Q để giảm liều thì liều mới không đáp ứng được
nồng độ thuốc trong huyết tương ở mức điều trị. Nếu giữ nguyên liều thì tại thời điểm
ngay sau khi đưa thuốc, nồng độ lại quá cao nhưng sau đó do khoảng cách quá dài nên
giai đoạn thuốc có nồng độ ở dưới mức điều trị cũng kéo dài, do đó hiệu quả điều trị thấp.
Những trường hợp này, ta có thể chọn 1 hệ số Q trung gian và dùng kết hợp cả 2 phương
pháp: vừa giảm liều vừa nới rộng khoảng cách.
Như vậy, xu hướng chung là theo dõi đáp ứng lâm sàng và giám sát nồng độ thuốc
trong máu để điều chỉnh liều. Ở nước ta hiện nay, hiện chưa thực hiện đo nồng độ thuốc
trong máu nên việc hiệu chỉnh liều sẽ khó khăn và căn cứ chủ yếu vào kinh nghiệm của bác sĩ điều trị.
Trong thực tế, mức liều cho bệnh nhân suy thận thường được nhà bào chế tính sẵn
và ghi rõ trong bảng hướng dẫn sử dụng thuốc. 64
Bảng 7.1. Hiệu chỉnh liều Ceftazidim theo độ thanh thải creatinin Clcr (ml/ph) Liều
Khoảng cách đưa thuốc 50-31 1g 12h/lần 30-16 1g 24h/lần 15-6 500mg 24h/lần <5 500mg 48h/lần Chú ý:
Nếu bệnh nhân suy thận nặng phải thẩm phân máu, thẩm phân phúc mạc thì quá
trình hiệu chỉnh liều còn phụ thuộc vào khả năng thuốc bị loại qua những đường này.
4. Một số nguyên tắc khi dùng thuốc ở bệnh nhân suy thận
Luôn dùng số thuốc cần thiết ở mức tối thiểu.
Cần tránh, nếu có thể, các thuốc gây độc cho thận.
Cần điều chỉnh liều dùng của nhiều loại thuốc cho bệnh nhân suy thận để tránh
nhiễm độc và đảm bảo hiệu quả của thuốc.
Dựa vào mức độ suy giảm chức năng thận để điều chỉnh liều thuốc. Mức độ điều
chỉnh này phụ thuộc vào mức độc hại của thuốc và khả năng thuốc đó được bài
xuất hoàn toàn qua thận hay được chuyển hoá một phần thành các chất chuyển hoá không hoạt tính.
Nhìn chung, tất cả bệnh nhân bị suy giảm chức năng thận có thể sẽ gặp nguy cơ
xấu khi được dùng thuốc với liều bằng với liều cho người có chức năng thận bình thường.
Điều chỉnh liều duy trì theo tình trạng lâm sàng. Có thể giảm liều duy trì bằng
cách giảm liều ở mỗi lần dùng mà không thay đổi khoảng cách đưa thuốc hoặc
giãn khoảng cách đưa thuốc mà không thay đổi liều.
Chức năng của thận (thể hiện ở mức lọc cầu thận, độ thanh thải creatinin) giảm
theo độ tuổi. Vì vậy, đối với người bệnh cao tuổi thì dùng thuốc với liều như liều
của bệnh nhân bị suy thận nhẹ.
Những thuốc cần tránh hoặc thận trọng khi sử dụng
Thuốc ức chế men chuyển angiotensin: captopril, enalapril, perindopril, quinapril
Aminoglycosid: amikacin, gentamicin, kanamycin, tobramycin 65
Thuốc chống ung thư: bleomycin, cyclophosphamid, cisplatin, methotrexat
Thuốc chẹn bêta: acebutolol, atenolol
Cephalosporin: cefadroxil, cefradin, cefazolin, cefuroxim, cefotaxim, ceftriaxon, ceftazidim
Thuốc chống viêm không steroid (NSAID): acid acetylsalicylic, diclofenac,
ibuprofen, indomethacin, ketoprofen, meloxicam, naproxen, piroxicam, tenoxicam
Penicilin: amoxicilin, ampicilin, benzylpenicilin
Quinolon: ciprofloxacin, acid nalidixic, norfloxacin, ofloxacin
Tetracyclin ngoại trừ doxycyclin và minocyclin Câu hỏi lượng giá
1. Tính độ thanh thải creatinin ở một bệnh nhân nam, 75 tuổi mắc bệnh đa u tủy biết rằng
nồng độ creatinine huyết thanh đo được sau 24 giờ là 2,1 mg/dl, nồng độ creatinine nước
tiểu là 50 mg/dl và lượng nước tiểu là 1400 ml.
2. Một bệnh nhân nữ 52 tuổi, cân nặng 65 kg nhiễm khuẩn với tụ cầu vàng kháng
methicillin (Methicillin-resistant Staphylococcus aureus: MRSA) cần được chỉ định dùng
vancomycin. Để tính toán được liều vancomycin, cần tính độ thanh thải creatinin (Clcr).
Biết bệnh nhân có nồng độ creatinin huyết thanh bằng (SCr) 1,8 mg/dl. Tính độ thanh thải
creatinin của bệnh nhân và độ thanh thải vancomycin (Clvan) với giả thiết độ thanh thải
vancomycin được tính bằng công thức sau: Clvan= 0,695 x Clcr (ml/phút/kg) + 0,05
3. Một bệnh nhân nam 70 tuổi, cân nặng 80 kg nhiễm khuẩn với trực khuẩn mủ xanh
(Pseudomonas aeruginosa) cần được chỉ định một liều tobramycin. Để tính toán được liều
tobramycin, cần biết độ thanh thải creatinine (Clcr). Biết nồng độ creatinine huyết thanh
(SCr) của bệnh nhân bằng 2,5 mg/dl. Tính độ thanh thải creatinine, hằng số tốc độ thải trừ
tobramycin (ke) và thời gian bán thải (t1/2) của tobramycin với giả thiết hằng số tốc độ thải
trừ ke (h-1)= 0,00293*Clcr (ml/phút) + 0,014 66
CHƯƠNG 3. GIÁM SÁT THUỐC ĐIỀU TRỊ (THERAPEUTIC DRUG MONITORING -TDM)
BÀI 8: PHƯƠNG PHÁP GIÁM SÁT THUỐC ĐIỀU TRỊ MỤC TIÊU
1. Trình bày được các phương pháp giám sát thuốc điều trị.
2. Nêu được các đối tượng chính của giám sát thuốc điều trị dựa vào nồng độ thuốc trong máu.
3. Phân tích được các yếu tồ cần cân nhắc khi diễn giải nồng độ thuốc. NỘI DUNG
Đáp ứng của thuốc khác nhau rõ rệt giữa các cá thể. Sự khác nhau này chủ yếu là do hai lý do chính:
1. Sự khác nhau về hấp thu, phân bố, chuyển hóa hoặc thải trừ (dược động học)
2. Sự khác nhau tại các receptor hoặc các đích tác dụng của thuốc khác (dược lực học)
Do đó, để đạt được mục tiêu điều trị, thuốc và liều lượng phải lựa chọn phù hợp
cho từng bệnh nhân. Trong quá trình điều trị, khâu giám sát thuốc điều trị về hiệu quả
điều trị, độc tính giúp tăng hay giảm liều cho phù hợp với mỗi bệnh nhân, giúp đạt hiệu
quả điều trị và giảm thiểu tác dụng phụ, độc tính của thuốc trên bệnh nhân.
1. Các phương pháp giám sát thuốc điều trị
Có 2 phương pháp chính được tiến hành để giám sát thuốc điều trị:
1.1. Phương pháp dược lực học
Đánh giá trực tiếp kết quả (mục tiêu điều trị). Ví dụ: theo dõi huyết áp của bệnh
nhân trong điều trị tăng huyết áp.
Dựa vào triệu chứng, dấu hiệu lâm sàng. Ví dụ: triệu chứng khô miệng do dùng liều atropin quá cao. 67
Bảng 8.1. TDM trên lâm sàng của một số thuốc Dấu hiệu cần Dấu hiệu Thuốc Bệnh/Tình trạng Tăng liều Giảm liều ngộ độc Desipramin Trầm cảm Còn trầm cảm Khô miệng, Hạ huyết áp mắt mờ khi đứng Furosemid
Phù trong suy tim, Vẫn còn phù
Mất cân bằng Hạ huyết áp, xơ gan, bệnh thận ion, da khô loạn nhịp tim Salicylat Thấp khớp Không
giảm Ù tai, buồn nôn Nhiễm toan viêm Thiopental
Gây mê phẫu thuật Không đủ mê Mê sâu Suy hô hấp
Dựa vào xét nghiệm cận lâm sàng. Ví dụ: làm xét nghiệm theo dõi thời gian
prothrombin (PT) khi dùng thuốc chống đông điều trị thuyên tắc mạch máu.
Bảng 8.2. TDM qua cận lâm sàng của một số thuốc Dấu hiệu cần Dấu hiệu Thuốc Bệnh Tăng liều Giảm liều ngộ độc Cyclosporin Ghép tạng Dấu hiệu thải Chức năng Suy thận cấp mô thận giảm L-thyroxin Suy giáp T3, T4 thấp T3, T4 tăng Cường giáp cao Pravastatin Cholesterol
máu Cholesterol còn SGOT, SGPT Transaminase cao cao tăng tăng, đau cơ Wafarin
Thuyên tắc mạch PT quá ngắn PT quá dài Xuất huyết máu
1.2. Phương pháp dược động học
Đối với một số thuốc, nồng độ thuốc trong huyết tương tỷ lệ tốt với đáp ứng dược
lý của thuốc đối với cơ thể. Vì vậy, người ta tiến hành đo gián tiếp nồng độ thuốc trong
máu để kiểm soát đáp ứng dược lý của thuốc, đặc biệt là đối với các thuốc có độc tính cao
hoặc liều điều trị gần với liều gây độc.
Giám sát thuốc điều trị theo phương pháp dược động học được định nghĩa là đo
nồng độ thuốc trong máu hay dịch cơ thể, dựa vào các thông số dược động học, để tính 68
toán tăng giảm liều dùng thuốc. Mục đích là đưa liều dùng thuốc trong máu cho phù hợp với khoảng điều trị.
Ví dụ: Điều chỉnh liều phenytoin ở bệnh nhân dựa trên nồng độ phenytoin trong huyết
thanh thay vì dựa trên số lần co giật giúp phòng được độc tính của thuốc.
2. Giám sát thuốc điều trị dựa vào nồng độ thuốc (Cp) 2.1. Khái niệm
Giám sát thuốc điều trị dựa vào nồng độ thuốc (kí hiệu TDM(Cp)): là một lĩnh vực
nghiên cứu của dược động học và hóa sinh lâm sàng, nhằm xác định và theo dõi nồng độ
thuốc trong máu tại một hoặc nhiều thời điểm khác nhau sau khi dùng thuốc, nhằm điều
chỉnh liều thuốc thích hợp cho từng bệnh nhân.
2.2. Đối tượngcần giám sát thuốc điều trị
Các thuốc cần theo dõi nồng độ thuốc điều trị có một số đặc điểm sau:
Các thuốc có cửa sổ điều trị hẹp (đối tượng chính) (ví dụ tỷ lệ nồng độ gây
độc/nồng độ có tác dụng < 2) như các thuốc chống co giật, thuốc tim mạch,
theophyllin, thuốc ức chế miễn dịch, thuốc chống trầm cảm ba vòng, thuốc
kháng virus, một vài loại kháng sinh và thuốc điều trị ung thư.
Các thuốc có độc tính cao, khi ngộ độc có nguy cơ gây nhập viện, tổn thương cơ
quan không hồi phục, thậm chí tử vong.
Các thuốc mà nồng độ thuốc trong máu chứ không phải liều dùng giúp tiên
lượng tốt hơn về đáp ứng điều trị của thuốc.
Các thuốc mà các thông số lâm sàng (theo dõi nhờ dược lực học) không được
định nghĩa rõ ràng để giúp điều chỉnh liều.
Các thuốc có mối liên hệ rõ ràng giữa nồng độ thuốc trong huyết tương với tác
dụng dược lý cũng như độc tính của thuốc. Với các thuốc gắn với protein mạnh
(tỷ lệ gắn trên 80%) thì nồng độ thuốc trong huyết tương dạng tự do tỷ lệ chặt
chẽ hơn với đáp ứng lâm sàng so với nồng độ thuốc toàn phần trong máu (phần
thuốc tự do + phần thuốc gắn protein), đặc biệt với những bệnh nhân tổn thương
gan, thận, người lớn tuổi, nên những thuốc này nên đo nồng độ thuốc tự do trong huyết tương.
Các thuốc điều trị thay thế cần được tối ưu (như thay thế hormon tuyến giáp thyroxine)
Các thuốc mà nồng độ thuốc trong huyết tương khác nhau nhiều giữa các cá
nhân khi dùng cùng một liều (ví dụ phenytoin) 69
Các thuốc cần thu thập dữ liệu về thuốc (hấp thu, chuyển hóa...), dữ liệu về các
thông số dược động học của thuốc.
Bảng 8.3. Danh sách một số thuốc cần giám sát nồng độ thuốc điều trị Nhóm thuốc Tên thuốc Thuốc chống co giật
Phenytoin, carbamazepin, acid valproic, phenobarbital,
primidon, ethosuximid, lamotrigine Thuốc tim mạch
Digoxin, quinidin, disopyramid, lidocain, procainamid Thuốc trị hen Theophyllin
Thuốc ức chế miễn dịch
Cyclosporin, tacrolimus, sirolimus, everolimus, acid mycophenolic Thuốc chống trầm cảm Amitriptylin, nortriptylin, doxepin, imipramin,
desipramin, clomipramin, trimipramin, lithium Thuốc kháng sinh
Amikacin, gentamicin, tobramycin, vancomycin Thuốc chống ung thư Methotrexat, cisplatin Thuốc kháng virus
Amprenavir, atazanavir, indinavir, lopinavir, saquinavir, nevirapin Thuốc giảm đau Acetaminophen, salicylat
Giám sát thuốc điều trị còn tiến hành trên những bệnh nhân có vấn đề về tuân thủ
điều trị (ví dụ: để kiểm tra sự tuân thủ điều trị dùng theophyllin trên bệnh nhân hen); hoặc
bị một số bệnh làm thay đổi các thông số dược động học như tổn thương gan, thận, bệnh
tim mạch, suy giáp, xơ nang; hoặc có khả năng gặp tương tác thuốc-thuốc, thuốc - thức ăn
nghiêm trọng (ví dụ: bệnh nhân đang dùng methotrexat khi dùng kèm với ciprofloxacin
có nguy cơ tăng nồng độ methotrexat).
Ngoài ra, giám sát thuốc điều trị còn được tiến hành trong trường hợp ngộ độc
thuốc như quá liều paracetamol, aspirin...
2.3. Những cân nhắc để diễn giải đúng nồng độ thuốc
a. Khoảng điều trị:
Một khoảng điều trị được định nghĩa là một khoảng nồng độ thuốc trong đó xác
suất đáp ứng lâm sàng mong muốn của thuốc cao và khả năng độc tính không thể chấp
nhận của thuốc thấp. Điều này có nghĩa rằng khoảng điều trị thực chất được thiết lập là
khoảng trung bình cho quần thể đó khi mà phần lớn bệnh nhân đáp ứng điều trị với tác 70
dụng phụ là chấp nhận được. Vì vậy, sẽ luôn có một số bệnh nhân cho thấy vẫn có tác
dụng điều trị tại nồng độ thuốc thấp hơn giới hạn tối thiểu, trong khi số khác sẽ bị độc
tính không thể chấp nhận được ngay tại nồng độ dưới giới hạn trên. Vì vậy, điều trị cho
một bệnh nhân luôn được chỉ dẫn bởi khoảng điều trị của cá nhân bệnh nhân đó.
Chỉ số trị liệu (therapeutic index = therapeutic ratio) = LD50 / ED50 trong đó LD50
là liều gây tử vong 50% quần thể (lethal dose 50), ED50 là liều có tác dụng trên 50% quần
thể (effective dose 50). Ví dụ: Diazepam có chỉ số trị liệu khoảng 100, digoxin 3.
Chỉ số bảo vệ (protective index) = TD50 / ED50 trong đó TD50 là liều gây độc cho 50% quần thể.
Hai chỉ số trên gần tương tự nhau. Chỉ số trị liệu và chỉ số bảo vệ càng lớn cho
thấy khoảng điều trị của thuốc đó càng rộng, thuốc đó càng an toàn. Ngược lại, chỉ số đó
càng nhỏ thì thuốc đó phải được điều chỉnh liều một cách cẩn thận và bệnh nhân cần được
theo dõi chặt chẽ các dấu hiệu ngộ độc.
Các yếu tố gì có thể ảnh hưởng đến khoảng điều trị trên một bệnh nhân: Bất cứ yếu
tố gì ảnh hưởng đến được động học của một thuốc sẽ ảnh hưởng đến khoảng điều trị.
Những yếu tố này bao gồm:
Chỉ định: Ví dụ, nồng độ digoxin trong huyết thanh cao hơn được yêu cầu để
điều trị rối loạn thất so với điều trị suy tim sung huyết. Nồng độ kháng sinh cao
hơn có thể được yêu cầu đối với các vi khuẩn đề kháng hoặc để xâm nhập vào
một số mô nhiễm khuẩn.
Chất chuyển hóa có hoạt tính: Sự có mặt của chất chuyển hóa có hoạt tính có
thể làm dịch chuyển khoảng điều trị cho bệnh nhân đó lên hoặc xuống.
Thuốc dùng kèm: Sự hiện diện của các thuốc khác có tác dụng dược lý tương tự
làm thay đổi khoảng điều trị. Tuổi bệnh nhân
Trạng thái điện giải: Hạ kali máu, hạ magie máu và tăng canxi máu làm tăng
hiệu quả tác dụng trên tim của digoxin và tăng độc tính của digoxin.
Bệnh đi kèm: Bệnh nhân có bệnh tim (bệnh xơ vữa động mạch vành) tăng nhạy
cảm với digoxin. Bệnh tuyến giáp cũng làm thay đổi đáp ứng với digoxin.
Tỷ số đối quang: Một số thuốc được dùng dạng hỗn hợp racemic các dạng đối
quang có thể có đặc tính đáp ứng/độc tính khác nhau. Vì vậy, một hỗn hợp với
tỷ lệ đối quang khác nhau sẽ cho đáp ứng/độc tính trên bệnh nhân khác nhau, ví dụ như disopyramid. 71
Khác nhau về kiểu gen: Các bằng chứng cho thấy đáp ứng với một số thuốc
được quyết định bởi kiểu gen. Với một thuốc đã được chọn, bệnh nhân có thể
xác định kiểu gen trước khi bắt đầu điều trị để xác định bệnh nhân là người đáp
ứng, người không đáp ứng hoặc người đáp ứng nhưng bị ngộ độc.
Khả năng gắn với protein huyết thanh: Theo lý thuyết, chỉ có nồng độ tự do của
thuốc trong máu mới có khả năng thiết lập trạng thái cân bằng với receptor
dược lý, vì vậy nồng độ tự do tiên lượng đáp ứng tốt hơn nồng độ toàn phần.
Hầu hết nồng độ thuốc trong huyết thanh, huyết tương, máu được đo dưới dạng
nồng độ tổng của dạng tự do và dạng không tự do (nồng độ toàn phần). Một số
bệnh nhân cho thấy độc tính trong khoảng điều trị thông thường có tỷ lệ gắn
với protein thấp bất thường và nồng độ thuốc tự do cao trong máu. Việc gắn
với protein của một thuốc thấp trong máu có thể là kết quả của hoặc giảm nồng
độ protein hoặc sự có mặt của các chất khác trong máu thay thế thuốc khỏi vị trí gắn protein.
Tóm lại, khoảng điều trị báo cáo bởi một phòng xét nghiệm chỉ là một hướng dẫn
ban đầu và không bảo đảm đáp ứng lâm sàng mong muốn trên bất cứ bệnh nhân cụ thể
nào. Các nỗ lực phải được tiến hành để xem xét các dấu hiệu đáp ứng lâm sàng và độc
tính khác, bổ sung cho việc đo nồng độ thuốc.
b. Thời điểm lấy mẫu:
Có hai cân nhắc chính về thời điểm lấy mẫu: (1) Phải đợi bao lâu sau khi dùng
thuốc hay điều chỉnh liều thuốc để lấy mẫu và (2) khi nào thì lấy mẫu trong khoảng cách dùng thuốc.
Đợi thuốc gần đạt đến trạng thái cân bằng
Sự phân bố của thuốc và vị trí của đích tác dụng của thuốc ảnh hưởng đến mối liên
hệ giữa nồng độ thuốc trong huyết tương và hiệu quả điều trị. Một tỷ lệ không đổi nồng
độ thuốc trong mô và nồng độ thuốc trong huyết tương chỉ đạt được trong giai đoạn cuối
của quá trình thải trừ thuốc. Vì vậy vào thời điểm đầu của khoảng cách đưa thuốc, nồng
độ thuốc trong huyết tương không phản ánh nồng độ thuốc trong khoảng mô gian bào một
cách chính xác. Do đó, đo nồng độ thuốc trong quá trình thuốc đang hấp thu hay phân bố
có thể cung cấp thông tin sai. Hình 8.1 minh họa trường hợp sử dụng digoxin. Đo nồng độ
thuốc cần phải được tiến hành tại thời điểm sau khi liều thuốc đã được phân bố. Do đó
cần thận trọng ghi thời điểm lấy mẫu để xác định nồng độ thuốc.
Nên đánh giá nồng độ thuốc khi gần với trạng thái cân bằng. Đối với thuốc tuân
theo động học bậc 1, thời gian để đạt trạng thái cần bằng có thể được suy đoán từ thời
gian bán thải t của thuốc. Đối với chế độ đưa liều cố định với khoảng cách đưa liều cố 1/2
định nên đợi tối thiểu 3t
trước khi lấy mẫu, thời gian này có thể 1/2 dài hơn trên một số 72
người bệnh do quá trình đào thải thuốc giảm. Trong trường hợp đưa liều không đều đặn,
nồng độ thuốc có thể đo tại cùng một thời điểm trong ngày có thể dùng để so sánh với nhau.
Hình 8.1. Đồ thị nồng độ thuốc trong huyết thanh-thời gian sau khi dùng digoxin
Có một số trường hợp ngoại lệ đối với quy tắc chung cần đợi cho đến khi trạng thái
ổn định đạt được trước khi lấy mẫu. Nếu nghi ngờ độc tính xuất hiện sớm, nồng độ thuốc
được đo ngay và có thể cần giảm tốc độ dùng thuốc ngay lập tức.
Thời điểm lấy mẫu trong khoảng cách dùng thuốc phụ thuộc vào mục đích muốn
xác định nồng độ đỉnh, nồng độ đáy, nồng độ trung bình ở trạng thái cân bằng hay nồng độ ngẫu nhiên
Đối với truyền thuốc với tốc độ không đổi, một khi trạng thái cân bằng đạt được,
nồng độ thuốc trong suốt quá trình truyền thuốc không đổi và mẫu để đo nồng độ thuốc có
thể lấy bất cứ thời điểm nào.
Đối với một thuốc đưa ngắt quãng, mặc dù trạng thái cân bằng đã đạt được, vẫn có
một sự dao động nồng độ thuốc nhất định. Nồng độ thấp nhất trong khoảng cách dùng
thuốc được gọi là nồng độ tối thiểu ở trạng thái cân bằng hay nồng độ đáy (Cmin). Nồng
độ cao nhất được gọi là nồng độ tối đa ở trạng thái cân bằng hay nồng độ đỉnh (Cmax).
Nồng độ trung bình ở trạng thái cân bằng (Css,avg) thể hiện nồng độ trung bình theo thời
gian trong suốt khoảng cách đưa thuốc.
Các thuốc với t1/2 khá dài được đưa dưới dạng chế phẩm giải phóng kéo dài hoặc
hấp thu chậm với dùng nhiều liều, nồng độ thuốc ổn định theo thời gian và có thể được đo 73
tại bất kì thời điểm nào trong khoảng thời gian dùng thuốc. Tuy nhiên, với thuốc giải
phóng tức thì t1/2 ngắn được dùng với khoảng cách dùng dài, sẽ có nhiều dao động hơn.
Biết thời gian dự kiến đạt nồng độ đỉnh của một chế phẩm là đặc biệt quan trọng đối với
những thuốc cho thấy dao động nhiều trong khoảng dùng thuốc. Trong trường hợp này,
dựa vào thời điểm lấy mẫu so với thời điểm dùng thuốc có thể đánh giá liệu nồng độ ghi
nhận được là gần với nồng độ đỉnh, nồng độ trung bình hay nồng độ đáy.
Sự lựa chọn thời điểm lấy mẫu trong khoảng cách đưa thuốc nên dựa trên câu hỏi
lâm sàng cần trả lời. Thường khi đưa liều lặp lại, một mẫu được lấy ngay trước thời điểm
dùng liều kế tiếp để xác định nồng độ đáy (Cmin) và một mẫu có thể lấy sau một khoảng
thời gian nhất định sau khi dùng thuốc (phụ thuộc vào thuốc đó) để xác định nồng độ đỉnh
(Cmax). Thông thường, nồng độ đáy được theo dõi cho hầu hết các thuốc, nhưng đối với
aminoglycoside và vancomycin, cả nồng độ đáy và nồng độ đỉnh đều được theo dõi.
Nồng độ đáy (Cmin) thường được dùng để khẳng định điều trị, đặc biệt nếu
khoảng điều trị được xây dựng dựa trên nồng độ đáy như đối với hầu hết các thuốc chống
động kinh. Nồng độ đáy cũng được khuyên dùng nếu chỉ định TDM là để tránh sự không
hiệu quả hoặc phân biệt không tuân thủ điều trị với thất bại điều trị. Trong đa số trường
hợp, nồng độ đáy được đo tại thời điểm ngay trước liều kế tiếp. Tuy nhiên, điều này
không luôn luôn đúng. Một số thuốc chế phẩm như giải phóng kéo dài (ví dụ acid
valproic bao tan ở ruột) được thiết kế để hấp thu ở ruột hơn là dạ dày. Do đó, thuốc không
bắt đầu hấp thu sau vài giờ sau khi dùng thuốc và nồng độ thuốc từ liều trước đó tiếp tục
giảm trong vài giờ trong khoảng cách dùng kế tiếp.
Nồng độ đỉnh (Cmax) được theo dõi ít thường xuyên hơn đối với các thuốc đường
uống bởi vì thời gian để đạt nồng độ đỉnh khó tiên lượng. Thời điểm lấy mẫu để đo nồng
độ đỉnh của một thuốc phụ thuộc vào đường dùng thuốc. Với tiêm tĩnh mạch, nồng độ
đỉnh có thể đạt được sau vài phút, trong khi đó thời gian để đạt nồng độ đỉnh sau khi dùng
viên nén theophyllin giải phóng kéo dài là 8 giờ. Nếu nồng độ đỉnh được yêu cầu, nên
xem tờ giấy hướng dẫn sử dụng để tham khảo thời gian đạt nồng độ đỉnh của chế phẩm
đó. Nồng độ đỉnh được theo dõi là thích hợp nếu bệnh nhân có triệu chứng ngộ độc tại
thời điểm tương ứng với nồng độ đỉnh. Nồng độ đỉnh có thể còn được dùng cho thuốc
dùng đường tĩnh mạch (aminoglycoside và chloramphenicol) bởi vì thời gian để đạt nồng
độ đỉnh tương ứng với thời điểm kết thúc việc truyền thuốc.
Đôi khi nồng độ trung bình của thuốc trong khoảng cách dùng thuốc (Css,avg)
được dùng để điều chỉnh liều. Theo dược động học, nồng độ trung bình bằng diện tích
dưới đường cong AUC trong suốt khoảng cách dùng thuốc (đòi hỏi đo nhiều mẫu) chia
cho khoảng cách dùng thuốc. Chú ý AUC trong khoảng cách dùng thuốc hoặc một phần
khoảng cách dùng thuốc được dùng như một thông số thay cho đo nồng độ thuốc chỉ đo
một lần đối với một vài thuốc như một số thuốc ức chế miễn dịch, thuốc chống ung thư 74
bởi vì nó cung cấp chỉ điểm cho toàn bộ lượng thuốc trong cơ thể. Tuy nhiên, xác định
AUC hoặc Css,avg bằng cách đo nhiều mẫu là không hiệu quả về kinh tế. Sau đây là cách
khác để ước lượng Css,avg mà không cần đo nhiều mẫu:
Lấy mẫu ở chính giữa thời điểm đạt Cmax và thời điểm đạt Cmin.
Đo nồng độ đáy Cmin và ước lượng nồng độ đỉnh Cmax như sau: Cmax = (Dose/Vd) + Cmin Cavg, ss = (Cmax +Cmin)/2
Nếu có rất ít sự dao động trong khoảng cách dùng thuốc, mẫu có thể được lấy bất
kì thời điểm nào trong khoảng cách dùng thuốc đều có thể được coi là nồng độ trung bình.
c. Mẫu, phương pháp thu thập mẫu, xét nghiệm:
Mẫu: Máu toàn phần, huyết tương và huyết thanh
Máu toàn phần, huyết tương và huyết thanh và dịch siêu lọc của huyết thanh là
những mẫu dùng phổ biến để đo nồng độ thuốc. Mức độ phân tử thuốc tự do trong máu
phân phối trong hồng cầu hoặc gắn với protein và nước của huyết tương phụ thuộc vào:
(1) khả năng phân phối vào trong hồng cầu, (2) nồng độ protein máu và (3) ái lực gắn protein của thuốc.
Mẫu huyết tương hoặc máu toàn phần được thu thập trong các ống nghiệm có chứa
chất chống đông. Huyết tương được tạo ra bằng cách ly tâm máu toàn phần đã chống
đông và thu thập lớp phía trên chứa huyết thanh, protein, thuốc tự do và thuốc gắn
protein. Mẫu huyết thanh được thu thập trong ống nghiệm không có chất chống đông và
các cục máu đông được hình thành sau đó được ly tâm. Mẫu huyết thanh thích hợp hơn để
đo nồng độ thuốc tự do. Huyết thanh được siêu lọc để tạo nước huyết thanh chỉ chứa
thuốc tự do. Nồng độ thuốc trong huyết thanh được siêu lọc luôn thấp hơn nồng độ thuốc
toàn phần, đặc biệt là các thuốc gắn mạnh với protein huyết thanh. Sự khác nhau duy nhất
của huyết thanh và huyết tương là trong huyết thanh, các yếu tố đông máu đã được sử
dụng. Nồng độ thuốc trong huyết thanh và huyết tương được xem là tương đương nhau.
Nồng độ thuốc trong máu toàn phần cao hơn nồng độ tương ứng trong huyết thanh hoặc
huyết tương nếu thuốc phân bố nhiều trong hồng cầu.
Chọn huyết thanh, huyết tương hay máu toàn phần để đo nồng độ thuốc phụ thuộc
vào yêu cầu của xét nghiệm sử dụng. Huyết thanh và huyết tương thích hợp hơn nếu
hemoglobin cản trở xét nghiệm. Máu toàn phần được dùng cho thuốc phân bố nhiều trong 75
hồng cầu như cyclosporine, tacrolimus. Một số phương pháp mới sử dụng máu toàn phần
mao mạch nên không cần ly tâm máu.
Kiến thức về tình trạng hoặc đặc tính bề ngoài của mẫu tại thời điểm xét nghiệm có
thể quan trọng. Một mẫu với dung dịch giống sữa có thể cho thấy lipid trong máu nhiều,
nếu mẫu có màu vàng tối đến vàng có thể cho thấy mức bilirubin cao, mẫu với màu hồng
nhạt có thể có sự tan máu. Tất cả các trường hợp này có thể dẫn đến làm tăng sai lệch kết
quả phân tích, phụ thuộc vào phương pháp đo. Những thuốc phân bố chủ yếu trong hồng
cầu hơn là huyết thanh sẽ cho kết quả nồng độ thuốc trong mẫu máu có tan máu cao hơn
thực tế trong huyết thanh, trong khi các thuốc phân bố ít hơn trong hồng cầu sẽ tan trong
huyết thanh hoặc huyết tương dẫn đến kết quả nồng độ thuốc trong trong mẫu máu có tan
máu thấp hơn thực tế trong huyết thanh.
Phương pháp thu thập mẫu
Chọn phương pháp thu thập mẫu máu cực kì quan trọng. Để phân tích huyết tương,
cần biết liệu yếu tố chống đông có cản trở xét nghiệm, ảnh hưởng đến độ ổn định và khả
năng gắn protein của thuốc hoặc thậm chí có thể làm loãng mẫu.
Các mẫu thay thế khác với mẫu máu
Nước bọt được xem là một mẫu thay thế không cần can thiệp thay cho mẫu máu,
đặc biệt ở trẻ em đối với một số thuốc. Nước bọt là một dịch siêu lọc tự nhiên của huyết
tương và vì vậy có thể phản ánh chặt chẽ hơn nồng độ thuốc tự do trong huyết thanh khi
so sánh với nồng độ thuốc toàn phần. Nồng độ thuốc trong nước bọt thấp hơn nhiều nồng
độ toàn phần trong huyết thanh của những thuốc gắn mạnh với protein, vì vậy cần chọn
xét nghiệm có độ nhạy cao.
Dùng nồng độ thuốc trong nước bọt như một thay thế cho nồng độ tự do hay toàn
phần trong huyết thanh hoặc huyết tương đòi hỏi tỷ lệ nồng độ thuốc trong nước bọt/huyết
tương phải ổn định, ít nhất ở bệnh nhân đó. Tỷ lệ này có thể bị ảnh hưởng bởi tốc độ tiết
của nước bọt, pH nước bọt và bị nhiễm với các dư lượng thuốc còn trong miệng.
Nước bọt toàn phần thường được thu thập và có thể được thu thập bằng biện pháp
kích thích hoặc không kích thích tạo nước bọt. Nước bọt được kích thích được ưa dùng
hơn: nó tạo một thể tích lớn hơn, giảm thiểu sự khác nhau giữa pH nước bọt và máu, cung
cấp tỷ số thuốc trong nước bọt/huyết tương ổn định hơn. Phương pháp kích thích tạo nước
bọt toàn phần bao gồm nhai kẹo cao su hoặc lăn một vật liệu trơ trong miệng hoặc dùng
một lượng nhỏ acid citric trên lưỡi.
Dịch nước mắt cũng là một giải pháp lý tưởng để đo nồng độ thuốc tự do vì pH của
nó khá ổn định. Nước mắt cũng có thể được kích thích bằng cách để mắt tiếp xúc với khói
thuốc lá. Một hạn chế chính của xét nghiệm thuốc trong nước mắt là độ nhạy của xét
nghiệm bởi vì thể tích nước mắt thu được không thể lớn. 76 Bảo quản
Các yếu tố sau có thể ảnh hưởng đến nồng độ thuốc trong mẫu: Ánh sáng, nhiệt
độ, vật liệu bảo quản, sự có mặt của các thuốc khác hoặc các chất nội sinh. Xét nghiệm
Diễn giải nồng độ của bất kỳ thuốc nào cũng đòi hỏi kiến thức đầy đủ về ưu nhược
điểm của phương pháp phân tích sử dụng. Các phương pháp phân tích phải được tiến
hành theo các tiêu chuẩn được chấp nhận để đạt độ chính xác và độ lặp lại, những đòi hỏi
về độ nhạy và độ đặc hiệu sẽ phụ thuộc vào mục đích sử dụng, khoảng điều trị, thể tích phân tích và loại mẫu.
Độ nhạy (sensitivity) của phương pháp với mẫu phân tích rất quan trọng để đưa ra
kết luận phân tích. Nếu phương pháp có độ nhạy cao, không phát hiện được có thể thực sự
nghĩa là không có thuốc trong mẫu. Nếu phương pháp có độ nhạy thấp, có thể thuốc trong
mẫu không đủ để được phát hiện. Giới hạn phát hiện tối thiểu sẽ là một thông tin rất quan
trong đối với các thuốc có khoảng điều trị thấp ví dụ với nồng độ mcg/l như digoxin. Các
xét nghiệm nhạy hơn được yêu cầu khi thể tích mẫu nhỏ như với trẻ sơ sinh. Nồng độ
thuốc trong nước bọt hoặc dịch siêu lọc của huyết thanh có thể đòi hỏi độ nhạy cao hơn
nếu những thuốc gắn mạnh với protein huyết thanh. Trong một số trường hợp, các xét
nghiệm có thể tiến hành đo nồng độ thuốc trong máu toàn phần đối với các thuốc phân bố
nhiều trong hồng cầu để khắc phục sự hạn chế về độ nhạy của phương pháp đo.
Độ đặc hiệu của phương pháp (specificity) cũng quan trọng. Nếu báo cáo nồng độ
thuốc từ một labo cao hơn mong đợi trong khi không có dấu hiệu ngộ độc trên bệnh nhân,
có thể nghi ngờ có một sự cản trở nào đó đã xảy ra trong xét nghiệm. Những chất có thể
cản trở xét nghiệm bao gồm các chất chuyển hóa của thuốc, các thuốc khác, các chất nội
sinh (như lipid, bilirubin, hemoglobin hoặc sản phẩm phụ tích lũy khi suy thận). Ví dụ,
các chất phản ứng miễn dịch giống digoxin nội sinh tích lũy trong huyết thanh của trẻ sơ
sinh, phụ nữ có thai và bệnh nhân bị bệnh gan, thận được ghi nhận làm tăng nồng độ digoxin giả tạo.
Sắc kí lỏng hiệu năng cao (HPLC) và sắc kí lỏng khí (GLC) vẫn được dùng trong
các labo lâm sàng và được xem là phương pháp chuẩn trong nhiều trường hợp. Xét
nghiệm miễn dịch đồng nhất, xét nghiệm miễn dịch phân cực huỳnh quang (FPIA), xét
nghiệm miễn dịch đa enzyme (EMIT) và xét nghiệm miễn dịch người cho enzyme vô tính
(CEDIA) cũng là những phương pháp được lựa chọn vì dễ sử dụng, khả năng tự động và
thời gian quay vòng nhanh. Những phương pháp miễn dịch thường đặc hiệu với các thuốc
gốc nhưng trong một số trường hợp, chất chuyển hóa hoặc các chất giống thuốc khác
cũng được nhận biết bởi kháng thể, gây ra sai số. Một vài thuốc không thích hợp cho xét
nghiệm miễn dịch. Lithium, một chất điện giải là một ví dụ và phải được phân tích dùng 77
kỹ thuật điện cực nhạy với ion (ISE), quang phổ hấp thụ phân tử hoặc phép đo quang phát
xạ ngọn lửa. Hầu hết các phương pháp xét nghiệm miễn dịch có khả năng cho kết quả trong vòng 1-5 phút.
d. Dùng nồng độ để điều chỉnh liều:
Cần chú ý cân nhắc trả lời các câu hỏi sau: liệu thuốc có tuân theo dược động học
bậc 1 (tuyến tính) tức tăng liều dùng hàng ngày sẽ tỷ lệ tuyến tính với tăng nồng độ
thuốc? Phương pháp điều chỉnh liều tập trung đạt nồng độ đỉnh và/hoặc đáy hay nồng độ trung bình mong muốn?
Chế độ liều dùng ngắt quãng có 3 thông số: tốc độ liều, khoảng cách dùng và liều.
Ví dụ một chế độ liều 240mg mỗi 8h, liều là 240mg, khoảng cách dùng là 8 giờ và tốc độ
liều là 720mg/ngày hoặc 30mg/giờ. Tốc độ liều là quan trọng bởi vì nó xác định nồng độ
trung bình (Css,avg) trong ngày. Mức độ dao động trong khoảng cách dùng bị ảnh hưởng
nhiều bởi khoảng cách dùng.
Điều chỉnh liều đối với thuốc tuân theo động học tuyến tính:
Nếu liều trung bình Css,avg được theo dõi thì Css,avg sẽ tăng tỷ lệ với tăng liều
dùng hàng ngày (tốc độ liều), bất kể khoảng cách dùng thuốc thế nào.
Nếu nồng độ đáy Cmin được theo dõi và khoảng cách dùng được giữ không đổi,
Cmin sẽ tăng tỷ lệ với liều hàng ngày.
Không nên thực hiện việc lấy mẫu sau khi điều chỉnh chế độ liều cho đến khi đạt
một trạng thái cân bằng mới. Đối với một thuốc với động học bậc 1, nên lấy mẫu cùng
một khoảng thời gian (tối thiểu sau 3t ) như sau khi dùng chế độ liều ban đầu. 1/2
Điều chỉnh liều đối với các thuốc không tuân theo động học tuyến tính:
Tất cả các thuốc sẽ cho thấy thải trừ không tuyến tính nếu nồng độ đủ cao được
dùng. Tuy nhiên, một số thuốc cho thấy tuân theo động học không tuyến tính (Michaelis-
Menten) ngay sau khi dùng thuốc. Điều này có nghĩa một sự tăng nhỏ tốc độ liều sẽ làm
tăng mạnh nồng độ thuốc trong huyết thanh. Tăng tốc độ liều sẽ đòi hỏi thời gian lâu hơn
để đạt trạng thái ổn định (ví dụ phenytoin).
Các phương pháp được miêu tả cho phép tiên đoán hiệu quả tăng tốc độ liều của
phenytoin ở quần thể trung bình hoặc đo thực tế các thông số xác định động học không
tuyến tính, là Vmax (tốc độ chuyển hóa tối đa) và Km (hằng số Michaelis) để điều chỉnh liều.
Phương pháp điều chỉnh liều dựa theo dược động học của quần thể hoặc phương
pháp điều chỉnh liều Bayesian, sử dụng xác suất thống kê, được thích dùng cho nhiều 78
trường hợp cá thể hóa điều trị. Chúng hữu ích cho những thuốc tuân theo động học tuyến
tính và không tuyến tính.
Câu hỏi lượng giá
1. Theo dõi nồng độ thuốc trong huyết thanh/huyết tương những thuốc nào sau đây được
xem là hữu ích bổ sung cho theo dõi lâm sàng: a) Carbimazole b) Warfarin c) Gentamicin d) Lithium e) Ciclosporin
2. Một bệnh nhân được chẩn đoán bị cơn co giật-co cứng (thể động kinh cơn lớn) được
khởi đầu điều trị bằng natri phenytoin 300mg/ngày. Bệnh nhân quay lại phòng khám sau
4 tuần và cho biết bệnh nhân không còn bị cơn động kinh nào kể từ khi dùng phenytoin.
Không có dấu hiệu hoặc triệu chứng của ngộ độc phenytoin. Tại sao cần đo nồng độ
phenytoin trong huyết thanh ở bệnh nhân này?
3. Bệnh nhân nữ M, 35 tuổi, người đang dùng aminoglycoside đơn trị để điều trị nhiễm
trùng gram âm. Dựa theo bệnh án, bệnh nhân nhận 5 liều tobramycin 100mg truyền trong
30 phút, cách nhau 8 giờ vào lúc 6 giờ sáng, 2 giờ chiều, 10 giờ chiều. Thời gian bán thải
của tobramycin, dựa theo độ thanh thải creatinin là 4,5h. Đo nồng độ tobramycin hai lần
được chỉ định để xác định liệu nồng độ đỉnh và đáy có nằm trong khoảng điều trị lần lượt
6-10 mg/l và 0,5-2 mg/l hay không. Đo lần 1: nồng độ đáy lúc 1:50 chiều, ngay trước lúc
truyền liều thứ 6 là 0,8 mg/l. Một nồng độ thứ hai được đo 30 phút sau khi kết thúc liều
thứ 6, được dùng để xác định Cmax là 7,8mg/l. Dựa trên thông tin này, chế độ liều được
duy trì. Đo lần 2: nồng độ được đo lặp lại 2 ngày sau, phát hiện Cmax là 10,1 mg/l và
Cmin là 2,9 mg/l. Chức năng thận dựa trên độ thanh thải creatinin không đổi. Nếu các
nồng độ thuốc đo lần 2 là chính xác thì cần điều chỉnh liều để tránh độc tính của thuốc.
Câu hỏi: Lí do nào có thể giải thích cho sự thay đổi nồng độ tobramycin huyết thanh?
Nồng độ tobramycin nào phản ánh chính xác chế độ liều hiện tại? 79
BÀI 9: GIÁM SÁT ĐIỀU TRỊ CỦA MỘT SỐ THUỐC MỤC TIÊU
1. Trình bày được các thông tin cần thiết để giám sát thuốc điều trị một số thuốc.
2. Áp dụng được các thông tin cần thiết để đo nồng độ thuốc phenytoin, digoxin và kháng sinh nhóm aminoglycosid. NỘI DUNG
1. Các thông tin cần thiết để theo dõi nồng độ thuốc Cp 1.1. Về bệnh nhân
Tình trạng lâm sàng:
Tuổi, giới tính, chiều cao, cân nặng, sắc tộc, có thai hay không.
Những bệnh đang có, đặc biệt các bệnh tim, gan, thận. Cận lâm sàng:
Chức năng thận (nồng độ creatinin, độ thanh thải creatinin).
Chức năng gan (prothombine time, albumin, bilirubin).
Mức liên kết protein (albumin, protein toàn phần trong huyết tương).
Các thông tin trên là cần thiết vì thông tin về bệnh mắc kèm, tình trạng sinh lý, thói
quen xã hội có thể giúp xác định ban đầu các thông số dược động học của quần thể đó,
để xác định liệu nồng độ đạt được có đúng như mong đợi hay không: Thông tin về
chức năng thận và albumin là quan trọng nếu nồng độ thuốc toàn phần được đo với một
thuốc gắn mạnh với protein huyết thanh; các chất nội sinh do bệnh gây ra có thể gây
cản trở xét nghiệm hay không; những bất thường về điện giải có thể ảnh hưởng đến
diễn giải một nồng độ đã cho (ví dụ digoxin).
Tiền sử đo nồng độ thuốc:
Ngày và thời gian của lần đo trước
Đáp ứng và chế độ liều thuốc liên quan đến nồng độ trước đó.
Cần biết là nồng độ nào của thuốc là hiệu quả, là gây độc tính,
nồng độ thuộc thay đổi thế nào khi thay đổi liều dùng 1.2. Về thuốc
Thuốc đang xem xét:
Loại thuốc, chế độ liều dùng, dạng dùng, đường dùng.
Thời gian dùng thuốc, thời điểm dùng liều cuối cùng
Sự tuân thủ trị liệu. 80
Bệnh nhân điều trị nội trú hay ngoại trú.
Thuốc có dùng chung: Tương tác thuốc.
Chất chuyển hóa của thuốc có hoạt tính hay không.
1.3. Về các thông số dược động học liên quan đến bệnh nhân Sinh khả dụng F. Hằng số hấp thu Ka.
Thể tích phân bố biểu kiến Vd.
Tỷ lệ thuốc không liên kết fu.
Độ thanh thải toàn phần ClT.
Độ thanh thải thận ClR. 1.4. Các xét nghiệm
Các test phải nhạy, chuyên biệt và chính xác.
Xét nghiệm miễn dịch đa enzyme EMIT (Enzyme multiplied immunoassay
technique): tính tự động cao, cho kết quả nhanh, tính nhạy trung bình, độ ổn định kém.
Xét nghiệm miễn dịch ELISA (Enzyme-Linked ImmunoSorbent Assay): tính tự
động cao, kết quả nhanh, tính nhạy trung bình.
RIA (Rich Internet Application): tính nhạy cao, cho kết quả chậm, nguy cơ nhiễm phóng xạ.
Xét nghiệm miễn dịch huỳnh quang FPIA (Fluorescence Plarizationo
Immunoassay): tính tự động cao, cho kết quả nhanh, kết quả ổn định, độ nhạy trung bình.
Sắc ký lỏng hiệu năng cao HPLC (High-performance liquid chromatography):
độ nhạy cao nhất, chi phí thấp nhất, tuy nhiên cho kết quả chậm và đòi hỏi
người được đào tạo sử dụng máy mới được sử dụng máy.
Kỹ thuật phân tích có tính đặc hiệu cao tránh được các hạn chế của các phương
pháp ít đặc hiệu hơn vì chúng phát hiện cả những chất liên quan như chất chuyển hóa của
thuốc. Thậm chí cả khi dùng các phương pháp đặc hiệu cao, người ta vẫn nhận thấy chúng
có thể cho các kết quả khác nhau. Mẫu báo cáo nên bao gồm xét nghiệm được dùng, các
thông số thể hiện độ đặc hiệu, độ nhạy và độ chính xác của phương pháp. 81
1.5. Thông tin về mẫu
Loại mẫu lấy để đo nồng độ thuốc: huyết tương, nước tiểu, dịch não tủy,
dịch tủy, sinh thiết, phân, hơi thở, nước bọt.
Thông tin về thời gian lấy mẫu so với lần cuối cùng dùng thuốc đặc biệt
quan trọng giúp xác định nồng độ đo được là gần Cmax, Cmin, hay Css.
Chất liệu ống lấy mẫu
Vị trí lấy mẫu so với vị trí đưa thuốc, nếu đường IV được dùng.
Loại nồng độ thuốc đo:
Nồng độ thuốc toàn phần. Nồng độ thuốc tự do.
Nồng độ chất chuyển hóa
Nồng độ thuốc nên được diễn giải khi cân nhắc đầy đủ thông tin trên. Biểu mẫu
yêu cầu đo nồng độ thuốc phải được thiết kế sao cho điền các thông tin quan trọng này.
Bảng 9.1. Các thông tin cần thiết khi giám sát điều trị một số thuốc Thuốc dùng Chất chuyển Thuốc Bệnh đi kèm Protein liên kết chung hóa hoạt động Gentamicin Bệnh thận - Penicillin - Cyclosporin Bệnh về miễn Mạnh Phenobarbital Vài chất hoạt dịch động Rifampicin Erythromycin Digoxin Bệnh thận - Thuốc lợi tiểu - Suy tim Nortryptillin Bệnh gan do Alpha- Quinidin 10-hydroxy uống rượu glycoprotein Nortryptillin Fluoxetin Tegretol Phenytoin Bệnh thận Albumin Valproic - Viêm gan mạn Cimetidin Theophyllin Viêm phổi Albumin Erythromycin COPD Phù phổi 82
2. Giám sát thuốc điều trị một số nhóm thuốc
2.1. Giám sát thuốc điều trị nhóm thuốc chống co giật
Các thuốc cần theo dõi gồm: phenytoin, carbamazepine, acid valproic,
phenobarbital, primidone, ethosuximide, lamotrigine. Phenytoin, carbamazepine và acid
valproic gắn mạnh với protein huyết tương. Do đó, nên đo nồng độ thuốc dạng tự do trong
huyết tương thay vì đo nồng độ toàn phần của thuốc, đặc biệt ở bệnh nhân có nồng độ
protein huyết tương thay đổi như suy gan, người lớn tuổi, phụ nữ có thai, bệnh nhân có
nồng độ albumin huyết tương thấp. Phenytoin
Đối với phenytoin, điều quan trọng cần lưu ý là phenytoin tuân theo dược động
học không tuyến tính và bệnh gan, thận, tình trạng có thai có thể ảnh hưởng lên sự phân
bố của thuốc. Cửa sổ điều trị của nồng độ phenytoin toàn phần trong huyết tương hay
huyết thanh là 10-20mg/l. Trong khi cửa sổ điều trị của nồng độ phenytoin dạng tự do là
1-2mg/l. Phenytoin gắn chủ yếu với albumin trong huyết tương. Tỷ lệ gắn giảm ở trẻ sơ
sinh, trẻ em, người lớn tuổi, bị bệnh gan, thận, xơ nang, bỏng, suy dinh dưỡng, AIDS.
Dùng đồng thời các thuốc gắn mạnh với albumin như acid valproic, aspirin, các thuốc
NSAID, các thuốc này đẩy phenytoin ra khỏi albumin làm nồng độ phenytoin tự do trong
huyết tương tăng. Vì vậy, ở những bệnh nhân này, với nồng độ phenytoin toàn phần có
thể nằm trong giới hạn bình thường 10-20mg/l, nhưng thực tế nồng độ phenytoin tự do
lớn hơn khoảng 1-2mg/l. Do đó, nếu dùng nồng độ phenytoin toàn phần thì sẽ không
chính xác. Vì vậy, để giải quyết vấn đề này có thể sử dụng một trong ba cách sau: (1) đo
nồng độ thuốc dạng tự do và so sánh với cửa sổ điều trị 1-2mg/l; (2) đo nồng độ toàn phần
và áp dụng công thức chuyển đổi để tính nồng độ thuốc dạng tự do (bằng cách nhân tỷ lệ
nồng độ thuốc tự do/nồng độ thuốc toàn phần trên bệnh nhân cùng tình trạng tương tự tra
trong tài liệu y khoa với nồng độ thuốc toàn phần đo được), sau đó so sánh với cửa sổ
điều trị 1-2mg/l; (3) đo nồng độ thuốc toàn phần và áp dụng công thức chuyển đổi để tính
nồng độ thuốc toàn phần hiệu chỉnh, sau đó so sánh với cửa sổ điều trị 10-20mg/l.
Công thức hiệu chỉnh áp dụng cho bệnh nhân hạ albumin máu và suy thận:
X = 0,2 khi albumin thấp và độ thanh thải creatinin ≥ 25ml/phút.
X = 0,1 khi albumin bình thường hoặc thấp và bệnh nhân bị thẩm tách máu. 83
Đối với bệnh nhân có độ thanh thải creatinin 10-25ml/phút không thể tính nồng độ
phenytoin toàn phần hiệu chỉnh chính xác. Vì vậy những bệnh nhân này cần được theo
dõi kỹ các dấu hiệu lâm sàng.
Trong trường hợp dùng cùng acid valproic, do acid valproic đẩy phenytoin ra khỏi
albumin, đồng thời ức chế chuyển hóa phenytoin nên nồng độ phenytoin toàn phần hiệu
chỉnh có thể tính theo công thức:
Nồng độ phenytoin toàn phần hiệu chỉnh = Nồng độ phenytoin toàn phần đo được +
(0.01 X nồng độ acid valproic X nồng độ phenytoin toàn phần đo được).
Thời gian lấy mẫu: Vì phenytoin tuân theo động học không tuyến tính nên thời
gian để thuốc đạt nồng độ cân bằng Css khó xác định. Thời gian bán thải t1/2 trung bình là
24 giờ. Nồng độ ổn định có thể chỉ đạt được sau 3 tuần. Một số nghiên cứu đề nghị nên
lấy mẫu sau 3-4 ngày (tức trước khi thuốc đạt Css) để bảo đảm nồng độ thuốc không tăng
quá nhanh. Có những công thức đã được xây dựng để tính toán Tmax khi đã biết Vmax và
Kmax. Điều lưu ý, là trái với động học tuyến tính (Tmax không phụ thuộc vào tốc độ
truyền thuốc), với động học không tuyến tính thì Tmax tăng khi tốc độ truyền thuốc tăng.
Người ta thường dùng nồng độ đáy Cmin phenytoin để theo dõi. Vì phenytoin tuân theo
động học không tuyến tính nên nồng độ thuốc trong máu trung bình trong một khoảng
cách đưa thuốc tăng nhanh hơn sự tăng tốc độ truyền thuốc.
2.2. Giám sát thuốc điều trị nhóm thuốc tác dụng trên tim mạch
Một số thuốc bao gồm digoxin, quinidin, disopyramid, lidocain, procainamid,
mexiletin, tocainid thường được giám sát thuốc điều trị vì mối liên hệ chặt chẽ giữa nồng
độ thuốc trong huyết tương và đáp ứng điều trị của thuốc. Do đó, độc tính của thuốc có
thể tránh được nếu thực hiện TDM. Digoxin là một trong những thuốc phổ biến nhất hay
dùng TDM. Disopyramid, lidocain, quinidin là những thuốc gắn mạnh với α1-
glycoprotein và nồng độ những thuốc này trong huyết tương ở dạng tự do thay đổi nhiều
ở những bệnh nhân có các bệnh lý khác nhau. Do đó, nên đo nồng độ thuốc tự do trong
máu thay vì nồng độ thuốc toàn phần của những thuốc này. Digoxin
Khoảng điều trị: Độc tính của digoxin đã được giảm rất nhiều từ khi thực hiện
TDM. Digoxin có tác dụng lên sự co thắt của cơ tim nên được dùng để điều trị suy tim
sung huyết, trong khi tác dụng lên nhịp tim được dùng để điều trị rối loạn nhĩ như rung
nhĩ, cuồng nhĩ và nhịp tim nhanh nhĩ kịch phát. Khoảng điều trị phổ biến là 0,5-2,0 mcg/l
ở người trưởng thành và 1-2,6 mcg/l ở trẻ sơ sinh. Khoảng điều trị thấp hơp (0,5-1 mcg/l)
thường được dùng để điều trị suy tim, có thể tăng lên 1,5 mcg/l. Khoảng điều trị cao hơn
(0,8-1,5 mcg/l) được yêu cầu để điều trị rối loạn nhịp nhĩ, có thể tăng lên đến 2 mcg/l. 84
Độc tính: 50% bệnh nhân có nồng độ digoxin trong huyết thanh trên 2,5 mcg/l cho
thấy độc tính của của digoxin. Các triệu chứng ngộ độc bao gồm yếu cơ, triệu chứng tiêu
hóa (chán ăn, nôn, buồn nôn, đau bụng và táo bón); triệu chứng trên thần kinh trung ương
(đau đầu, mất ngủ, lơ mơ, chóng mặt và thay đổi màu sắc khi nhìn), các triệu chứng
nghiêm trọng trên tim (chậm nhịp tim tâm nhĩ thất độ 2, độ 3; co thất sớm và nhanh nhịp
thất). Trong thực tế, nhiều triệu chứng của rối loạn nhịp tim gặp phải khi dùng nồng độ
digoxin cao rất giống với triệu chứng bệnh được điều trị, vì vậy cần theo dõi nồng độ
thuốc điều trị digoxin để phân biệt độc tính với dấu hiệu không đáp ứng điều trị.
Có một số trường hợp sinh - bệnh lý có thể làm chuyển dịch khoảng điều trị của
digoxin. Độc tính của digoxin có thể dễ xảy ra hơn khi bệnh nhân hạ kali, magie, calci
máu hoặc bị các bệnh tim như bệnh tim xơ vữa động mạch vành hoặc nhồi máu cơ tim cũ.
Bệnh nhân bị cường giáp thường đề kháng với digoxin hơn.
Chỉ định: Những chỉ định chủ yếu của theo dõi nồng độ digoxin bao gồm: nghi ngờ
ngộ độc digoxin để xác định lượng thuốc giải độc dùng kháng thể gắn đặc hiệu với
digoxin (mảnh Fab gắn digoxin - Digibind®); nghi ngờ ngộ độc do tiêu hóa cây cỏ có
chứa các glycoside có cấu trúc tương tự; chức năng thận giảm đòi hỏi phải điều chỉnh tốc
độ đưa thuốc; nghi ngờ có tương tác thuốc như với thuốc kháng acid, amiodarone, thuốc
kháng sinh đường uống, cholestyramin, cyclosporin, metoclopramid, neomycin, quinidin,
spironolacton, sulfasalazin, verapamil.
Thời điểm lấy mẫu: Thời gian bán thải trung bình của digoxin ở người trưởng
thành với chức năng thận bình thường xấp xỉ 2 ngày và cần ít nhất 7 ngày để đạt trạng
thái cân bằng. Trong trường hợp điều trị quá liều digoxin bằng kháng thể đặc hiệu với
digoxin, mẫu máu để đo nồng độ digoxin trong huyết thanh không nên lấy sớm hơn 10
ngày sau khi dùng kháng thể. Bởi vì hầu hết các đo lường bằng phản ứng miễn dịch có thể
xác định cả digoxin dạng tự do và dạng gắn với kháng thể, việc lấy mẫu sớm có thể dẫn
đến kết quả nồng độ digoxin cao giả tạo.
Mẫu lấy trong khi thuốc đang hấp thu và phân phối cũng không thích hợp để so
sánh với khoảng điều trị. Nồng độ digoxin trong máu không phản ánh mức độ quan trọng
hơn nồng độ digoxin trong mô cơ tim nếu đo trong vòng 6 giờ sau khi dùng thuốc (thậm
chí là 12 giờ). Mẫu máu nên được lấy giữa 6 giờ sau khi dùng thuốc và ngay trước liều kế
tiếp. Thời điểm lấy mẫu digoxin không thích hợp là vấn đề chính ở hầu hết các bệnh viện.
Mẫu, phương pháp thu thập mẫu và xét nghiệm: Huyết thanh hoặc huyết tương
được chống đông bằng heparin hoặc EDTA có thể được dùng. Dịch siêu lọc huyết thanh
được dùng để xác định nồng độ digoxin tự do ở bệnh nhân điều trị với mảnh Fab
(Digibind®). Mẫu ổn định trong 24 giờ ở 2°C và 1-2 tuần ở -20°C. Nồng độ digoxin trong
nước bọt được đo trong một số nghiên cứu, nhưng không cho thấy mối liên hệ đủ mạnh
với nồng độ tự do cũng như toàn phần trong huyết thanh. 85
Nồng độ digoxin huyết thanh được đo chủ yếu bằng phương pháp miễn dịch.
Kháng thể digoxin trong các phản ứng miễn dịch này phản ứng chéo với các chất chuyển
hóa digoxin, các chất miễn dịch giống digoxin nội sinh (DLIS) và các thuốc khác cùng
chất chuyển hóa của chúng (spironolactone và chất chuyển hóa có hoạt tính của nó
canrenone, digitoxin và chất chuyển hóa của digitoxin). Cần chú ý các cản trở này có thể
gây tăng hoặc giảm nồng độ digoxin giả tạo. Các chất chuyển hóa digoxin gây cản trở xét
nghiệm tích lũy ở bệnh nhân bị bệnh thận. DLIS chủ yếu trong máu bệnh nhân bị bệnh
gan và thận, người mang thai hay trẻ sơ sinh. Do có sự sai lệch kết quả phân tích, việc
theo dõi các dấu hiệu và triệu chứng lâm sàng bổ sung cho nồng độ thuốc trong huyết thanh là rất quan trọng.
Nguyên nhân chính gây cản trở đo nồng độ digoxin trong huyết thanh bằng phản
ứng miễn dịch là sự có mặt của mảnh Fab (Digibind) dùng như một chất giải độc khi ngộ
độc digoxin. Kháng thể digoxin từ phản ứng miễn dịch gây cản trở này bằng cách cạnh
tranh với mảnh Fab của digoxin trong cùng một mẫu. Ở những bệnh nhân suy thận, sự
cản trở này kéo dài hơn 10 ngày sau khi dùng thuốc giải độc. Siêu lọc huyết thanh loại bỏ
mảnh Fab gắn digoxin cho phép đo nồng độ digoxin tự do khá tin cậy.
Dùng nồng độ để điều chỉnh liều: Điều chỉnh liều digoxin dựa trên nồng độ
digoxin trong huyết thanh là dễ làm. Bởi vì thuốc tuân theo động học bậc 1 (tuyến tính),
một sự tăng liều hàng ngày digoxin sẽ làm tăng tỷ lệ tương ứng nồng độ thuốc trong huyết
thanh tại thời điểm đó trong khoảng cách dùng. Nồng độ huyết thanh được dùng để điều
chỉnh liều được đo không sớm hơn 6-8 giờ sau khi dùng liều cuối cùng.
Khả năng gắn protein, chất chuyển hóa có hoạt tính và các cân nhắc khác:
Digoxin chỉ có 20-30% gắn với protein huyết thanh. Vì vậy, nồng độ toàn phần trong
huyết thanh sẽ phản ánh nồng độ tự do. Hoạt tính sinh học của chất chuyển hóa của
digoxin là khiêm tốn so với thuốc gốc và sự có mặt của chất chuyển hóa không ảnh hưởng
đến diễn giải nồng độ digoxin huyết thanh.
2.3. Giám sát thuốc điều trị nhóm thuốc kháng sinh
Các kháng sinh hay thực hiện TDM là amikacin, gentamicin, tobramycin,
vancomycin, cefazolin, ciprofloxacin.
Nhóm kháng sinh aminoglycosid
Kháng sinh aminoglycoside (AG) chủ yếu được tiêm tĩnh mạch để điều trị nhiễm
trực khuẩn gram âm đề kháng với các kháng sinh ít độc hơn. Tuy nhiên, thuốc lại có độc
tính cao trên thận và tai.
Mối liên quan giữa nồng độ - độc tính: Độc tính trên thận và tai là tác dụng có hại
phổ biến nhất của aminoglycosid. Độc tính trên tai dường như liên quan đến thời gian 86
dùng thuốc kéo dài (hơn 7-10 ngày) với Cmax trên 12-14mg/l đối với gentamicin và
tobramycin, trên 35-40 mg/l với amikacin. Bệnh nhân với Cmin trên 2-3mg/l (gentamicin,
tobramycin) hoặc 10mg/l (amikacin) trong một thời gian dài tăng nguy cơ bị độc trên thận.
Mối liên hệ giữa nồng độ - hiệu quả: Hai đặc tính dược lực học của nhóm
aminoglycosid là tính diệt khuẩn phụ thuộc nồng độ và tác dụng "hậu kháng sinh". AG
diệt vi khuẩn nhanh nhất khi nồng độ của chúng lớn hơn nồng độ ức chế tối thiểu (MIC)
của vi khuẩn và tác dụng này phụ thuộc vào nồng độ thuốc. Thuốc còn thể hiện tác dụng
"hậu kháng sinh" (post-antibiotic) tức tác dụng diệt khuẩn vẫn tiếp tục thậm chí sau khi
nồng độ đáy Cmin thấp dưới nồng độ ức chế tối thiểu (MIC). Vì tác dụng diệt khuẩn phụ
thuộc nồng độ và tác dụng hậu kháng sinh của AG giải thích tại sao chế độ liều với
khoảng cách liều kéo dài (KCLKD) cho thấy an toàn và hiệu quả trên nhiều bệnh nhân.
Chế độ liều truyền thống (TT) là dùng AG nhiều lần/ngày còn chế độ liều KCLKD nghĩa
là tăng liều thuốc và tăng khoảng cách dùng thuốc ví dụ dùng 1 liều mỗi 24, 36 hay 48
giờ. Vì vậy, tỷ số Cmax/MIC là một chỉ số tiên lượng hiệu quả của thuốc. Tỷ lệ này nên
đạt ít nhất 8-10 để AG phát huy hoạt tính diệt khuẩn.
Khoảng điều trị: Khoảng điều trị cho nồng độ đáy và đỉnh được thiết lập với AG
và chỉ áp dụng cho chế độ liều TT (nhiều lần trong ngày). Đối với gentamicin và
tobramycin, khoảng điều trị Cmax là 6-10 mg/l và Cmin là 0,5-2 mg/l. Đối với amikacin
Cmax là 20-30 mg/l và Cmin là 1-8 mg/l (xấp xỉ 4 lần MIC). Không có khoảng điều trị
đối với chế độ liều KCLKD, liều thuốc được đưa để đạt nồng độ đỉnh xấp xỉ 10 lần MIC
và nồng độ đáy không phát hiện được trong vòng 4 giờ trước khi tiêm liều tiếp theo. Đôi
khi, nồng độ huyết thanh được lấy sau khi truyền liều thuốc được dùng chỉ để điều chỉnh
khoảng cách liều, không dùng để kiểm tra hiệu quả và độc tính.
Thời gian lấy mẫu: Đối với chế độ liều KCLKD với chức năng thận bình thường,
trạng thái cân bằng không bao giờ đạt được vì mỗi liều đều bị loại sạch trước khi dùng
liều tiếp theo. Phương pháp phát triển bởi Nicolau và cộng sự (gọi là phương pháp
Hartford) đòi hỏi một mẫu máu duy nhất đo giữa 6 và 14 giờ sau khi kết thúc truyền
thuốc. Mẫu này được xem là mẫu ngẫu nhiên nhưng thời điểm thu thập mẫu phải được
ghi nhận. Nồng độ này được dùng đối chiếu theo một biểu đồ để xác định liệu các khoảng
cách dùng thuốc khác nhau có nên dùng. Nồng độ quá cao, dựa theo biểu đồ, sẽ cho thấy
thuốc đó không bị loại sạch như tiên đoán ban đầu, nên đề nghị kéo dài khoảng cách dùng
thuốc. Đối với chế độ liều TT, cần phải đợi cho đến khi trạng thái cân bằng đạt được
trước khi lấy mẫu. Thời gian bán thải của AG là 1,5-3 giờ đối với người trưởng thành có
chức năng thận bình thường nhưng có thể kéo dài 72 giờ ở bệnh nhân suy thận nghiêm
trọng. Bởi vì khoảng cách liều của AG là thường được điều chỉnh gấp 2-3 lần thời gian
bán thải của thuốc đó, vì vậy nguyên tắc chung là trạng thái cân bằng đạt được sau 3 đến 87
4 liều thuốc. Một số bệnh nhân có thể lấy mẫu ngay lập tức sau khi dùng liều đầu tiên để
xác định thông số dược động học cho mục đích cá nhân hóa chế độ liều. Điều này xảy ra
với những bệnh nhân tiên lượng sẽ có thông số dược động học thay đổi, như những người
ở khoa cấp cứu, những người đòi hỏi điều trị ngay lập tức bởi vì nhiễm khuẩn nguy hiểm đến tính mạng.
Mẫu, phương pháp thu thập mẫu và xét nghiệm:
Huyết thanh hoặc huyết tương được chống đông bằng EDTA. Heparin cản trở một
số xét nghiệm nên khuyến cáo không nên dùng để chống đông trừ khi labo đã loại trừ
nguy cơ này. Một nghiên cứu cho thấy nồng độ gentamicin trong nước bọt tiết ra bằng
kích thích bởi acid citric ở trẻ em là một chỉ điểm tốt cho nồng độ gentamicin đáy trong
huyết tương, nhưng chỉ dùng khi chế độ liều KCLKD.
Huyết thanh hay huyết tương nên được phân tích trong vòng 2 giờ lấy mẫu hoặc
làm lạnh tại 0-5°C. Điều này cực kì quan trọng với các mẫu chứa kháng sinh beta-lactam
như penicillin D, ampicillin, carbenicillin hoặc ticarcillin. Các kháng sinh beta-lactam
thường dùng chung với AG, gắn vật lý với AG trong máu dẫn đến bất hoạt chúng. Trong
in vivo, điều này có nghĩa AG được loại sạch nhanh hơn bình thường. Quá trình bất hoạt
AG tiếp tục có thể xuất hiện sau khi thu thập mẫu.Ví dụ, nồng độ huyết thanh là 7 mg/l tại
thời điểm thu thập mẫu có thể trở thành 6 mg/l sau một thời gian để ở nhiệt độ phòng.
Dùng nồng độ huyết thanh thấp giả tạo này có thể dẫn đến lỗi khi xác định các thông số
dược động học của AG. Nếu xét nghiệm ngay lập tức không thể, huyết thanh hoặc huyết
tương nên được làm lạnh ngay.
Xác định liều đầu tiên:
Trước khi đo nồng độ thuốc trong huyết thanh, chỉ dựa trên các thông số trên quần
thể để xác định liều khởi đầu cho bệnh nhân. Phương pháp đưa liều của Sarrubi và Hull
dựa trên độ thanh thải của creatinine để xác định liều đầu tiên dựa trên một toán đồ cho
sẵn. Tuy nhiên, phương pháp này nên được xem như một chỉ dẫn ban đầu và nên dựa vào
nồng độ thuốc trong huyết thanh và đáp ứng lâm sàng để điều chỉnh liều tiếp theo.
Xác định cân nặng tính liều (dosing weight -DW)
Tính trọng lượng nạc cơ thể (Lean Body Weight - LBW)
LBW (nam) = (1,10 x Cân nặng (kg)) - 128 (Cân nặng2/(100 x Chiều cao (m))2)
LBW (nữ) = (1,07 x Cân nặng (kg)) - 148 ( Cân nặng2/(100 x Chiều cao (m))2)
DW = LBW + ((ABW - LBW) x CF
Trong đó ABW (actual body weight) là cân nặng thực của bệnh nhân 88
CF là hệ số điều chỉnh cho người béo phì, thường là 40% nhưng giá trị là khác nhau tùy
thuốc: amikacin: 38%; gentamicin: 43% ; kanamycin: 0% ; neltimicin: 50% ; tobramycin: 58%.
Xác định liều tấn công (loading dose- LD)
Gentamicin, Tobramycin, Neltimicin: LD = 2mg/kg x DW
Amikacin, Kanamycin: LD = 7,5mg/kg x DW
Xác định liều duy trì (maintenance dose -MD)
Ước lượng hằng số tốc độ thải trừ (ke): ke = 0,01 + (Clcr x 0,0024)
Trong đó Clcr là độ thanh thải creatinin.
Ước lượng thể tích phân bố (Vd): Vd = 0,27 l/kg x DW
Tính liều duy trì lý tưởng (ideal maintenance dose -IMD):
IMD = ke x Vd x Cmax x (1 - e -ke x τ )/(1- e -ke x tinf)
Trong đó: τ là khoảng cách liều lý tưởng; tinf là thời gian truyền; Cmax là nồng độ đỉnh cần đạt
Chọn liều và khoảng cách liều thích hợp
Tính nồng độ đỉnh và đáy dự đoán:
Nồng độ đỉnh = (MD/tinf x Ví dụ x ke) x (1 - e -ke x tinf )/(1- e -ke x τ)
Nồng độ đáy = Nồng độ đỉnh x e -ke x (τ - tinf)
Dùng nồng độ để điều chỉnh liều: Phương pháp dùng liều KCLKD được dùng để
lợi dụng ưu việt của tác dụng diệt khuẩn phụ thuộc liều và tác dụng hậu kháng sinh của
AG. Phương pháp Hartford liên quan với đưa một liều mg/kg để đạt nồng độ đỉnh xấp xỉ
10 lần MIC. Sau đó, một mẫu lấy giữa 6-14 giờ sau khi kết thúc truyền thuốc và so sánh
với một biểu đồ cho sẵn, nó sẽ chỉ ra khoảng cách liều duy trì thích hợp, thường 24, 36
hoặc 48 giờ. Phương pháp đưa liều KCLKD không được khuyến cáo dùng cho một số
bệnh nhân, bao gồm người lớn tuổi có độ thanh thải creatinin dưới 30 ml/phút, lọc máu,
có thai, xơ nang, giảm bạch cầu trung tính, trẻ sơ sinh, tiền sử rối loạn chức năng tiền
đình/nghe, nhiễm khuẩn gram dương (khi AG dược dùng phối hợp với kháng sinh khác),
nhiễm khuẩn do vi khuẩn hiếm khí gram dương hình que (mycobacteria) những người
viêm nội tâm mạc do vi khuẩn đường ruột, suy thận, viêm màng não, viêm tủy xương hoặc bỏng.
Khả năng gắn protein, chất chuyển hóa có hoạt tính và các cân nhắc khác: Dưới
10% AG gắn với protein huyết thanh và nồng độ tự do sẽ luôn phản ánh nồng độ toàn
phần trong huyết thanh. Chất chuyển hóa của AG không có hoạt tính. 89
Chỉ định: Giám sát thuốc điều trị đối với aminoglycoside rất cần thiết ở những
bệnh nhân với nhiễm khuẩn nghiêm trọng được điều trị kéo dài, đặc biệt nếu có sự thay
đổi các thông số dược động học (như suy thận, bỏng, xơ nang, tuổi cao, có thai, nhiễm
khuẩn huyết) và nguy cơ cao gặp độc tính (như ở bệnh nhân dùng đồng thời thuốc lợi tiểu
quai hoặc thuốc độc cho thận như amphotericin, cyclosporin, vancomycin).
Bảng 9.2. Giám sát điều trị một số thuốc Thuốc Mẫu máu
Cửa sổ điều trị (µg/ml) Phenytoin HTh hoặc HTg 10-20 Carbamazepine HTh 4-12 Acid valproic HTh hoặc HTg 50-100 Phenobarbital HTh hoặc HTg 15-40 Digoxin HTh hoặc HTg 0,8-2,0 Disopyramide HTh hoặc HTg 1,5-5,0 Lidocaine HTh hoặc HTg 1,5-5,0 Quinidine HTh hoặc HTg 2-5 Theophyllin HTh hoặc HTg 10-20 Cyclosporin Máu toàn phần 0,15 – 0,4 Tacrolimus Máu toàn phần 0,005-0,015 Acid mycophenolic HTh hoặc HTg 1-1.3 Amikacin HTh hoặc HTg Cmin <5; Cmax = 12 - 25 Gentamicin HTh hoặc HTg Cmin =1-2; Cmax=4-8 Tobramycin HTh hoặc HTg Cmin=1-2; Cmax=4-8 Vancomycin HTh hoặc HTg Cmin=5-15; Cmax=30-40 Cefazolin HTh hoặc HTg Cmax=60-120 Ciprofloxacin HTh hoặc HTg Cmin=0,5-2; Cmax=3-5
HTh: Huyết thanh; HTg: Huyết tương 90 Câu hỏi lượng giá
1. Một bệnh nhân nam 66 tuổi, được nhập viện vào khoa cấp cứu với triệu chứng buồn
nôn, tiêu chảy, hoa mắt, dáng đi không thăng bằng. Bệnh nhân được chẩn đoán động kinh
cách đây 3 năm và dùng thuốc chống động kinh đường uống tại nhà: acid valproic 1000
mg X 2 lần/ngày, phenytoin natri 200 mg X 3 lần/ngày, carbamazepine 300 mg X 2
lần/ngày và levetiracetam 400 mg lúc đi ngủ. Bệnh nhân có tiền tử cao huyết áp, mỡ máu
cao và đang dùng aspirin 81mg hàng ngày, simvastatin 40 mg lúc đi ngủ và metoprolol
100 mg X 2 lần/ngày. Kiểm tra bệnh nhân phát hiện chứng giật cầu mắt hai bên, mất điều
hòa. Dấu hiệu lâm sàng dường như cho thấy ngộ độc thuốc và đo nồng độ thuốc toàn phần trong huyết thanh:
Carbamazepine: 6,4 mg/l ( khoảng điều trị: 4-12 mg/l)
Phenytoin: 9,3 mg/l (khoảng điều trị 10-20 mg/l)
Acid valproic: 72 mg/l (khoảng điều trị 50-100 mg/l)
Nồng độ tự do của ba thuốc trên cũng đo được:
Carbazepine: 1,9 mg/l (khoảng điều trị 1-3 mg/l)
Phenytoin: 1,3 mg/l (khoảng điều trị 1-2 mg/l)
Acid valproic: 13,4 mg/l (khoảng điều trị 2,5-10 mg/l)
Acid valproic được dừng 24 giờ sau đó dùng lại với liều 250mg X 3 lần/ngày. Các triệu
chứng biến mất trong vòng 24 giờ. Nồng độ tự do acid valproic được đo một tuần sau đó là 5,2 mg/l.
Câu hỏi: Nồng độ thuốc chống động kinh toàn phần ở bệnh nhân là thông số không chính
xác so với nồng độ tự do để điều chỉnh liều?
2. Một bệnh nhân với hạ albumin máu được khởi đầu điều trị bằng acid valproic để điều
trị động kinh cơn nhỏ (cơn vắng ý thức). Nồng độ acid valproic trong huyết thanh được
đo là 75mg/l. Bệnh nhân cho biết cô ấy không trải qua bất cứ cơn động kinh nào từ khi
bắt đầu điều trị, nhưng cô ấy cảm thấy lơ mơ hàng ngày. Xét nghiệm thấy nồng độ
albumin huyết thanh là 2,8 g/dl (bình thường là 3,5-5 g/dl). Hãy diễn giải nồng độ acid valproic? 91
TÀI LIỆU THAM KHẢO Tiếng Việt
1. Bộ môn Bào chế trường Đại học Dược Hà Nội (2010), Kỹ thuật bào chế và sinh
dược học các dạng thuốc, tập 1 và 2, NXB Y học.
2. Bộ môn Dược lâm sàng trường Đại học Dược Hà Nội (2011), Dược lâm sàng, NXB Y học.
3. Bộ y tế (2011), Dược thư quốc gia Việt Nam.
4. Trần Thị Thu Hằng (2009), Dược động học lâm sàng, NXB Phương Đông. Tiếng Anh
1. Arthur J. Atkinson et al. (2007), Principles of clinical pharmacology 2nd ed, Elsevier.
2. Bertram G.Katzung (2004), Basic & Clinical Pharmacology 9th Ed., Mc GrawHill.
3. John E. Murphy, Pharm.D., FASHP , FCCP (2008), Clinical Pharmacokinetics, 4th ed., ASHP.
4. Joseph T. Dipiro et al. (2008), Pharmacotherapy: A pathophysiologic approach. 7th ed, McGrawHill.
5. Larry A. Bauer (2008), Applied clinical pharmacokinetics. 2nd ed., McGrawHill.
6. Laurence L. Brunton, Keith L. Parker. (2008), Goodman and Gilman’s Manual of
pharmacology and therapeutics, McGrawHill.
7. Malcohm Rowland (1995), Clinical Pharmacokinetics, 3th ed., Lippincott William & Wilkins.
8. Michael E. Winter (2004), Basic Clinical Pharmacokinetics, 4th ed., Lippincott William & Wilkins. 92 PHỤ LỤC PHỤ LỤC 1
Bảng 1. Các thông số dược động học của một số thuốc thường dùng Liên hợp Tên thuốc SKD (F)
với protein Cl (ml/phút) Vd (l) t1/2 (h) (%) Acetaminophen 88 ± 15 0-15 350 ± 100 67 ± 8 2 ± 0,4 Acyclovir 15 – 30 15 ± 4 330 ± 80 50 ± 17 2,4 ± 0,7 Amoxicillin 93 ± 10 18 370 ± 90 29 ± 13 1 ± 0,1 Ampicillin 62 ± 17 18 ± 2 270 ± 50 20 ± 5 1,3 ± 0,2 Aspirin 68 ± 3 49 650 ± 80 11 ± 2 0,25 ± 0,3 Atenolol 56 ± 30 <5 97 39 ± 20 6,3 ± 1,8 Catopril 65 30 890 ± 210 49 ± 6 1,9 ± 0,5 Carbamazepine >70 74 ± 3 89 ± 37 98 ± 26 15 ± 5 Cephalexin 90 ± 9 14 ± 3 300 ± 80 18 ± 2 0,9 ± 0,18 Chloramphenicol 75 – 90 53 ± 5 170 ± 14 66 ± 4 2,7 ± 0,8 Chlordiazepoxide 100 96,5 ± 1,8 38 ± 34 21 ± 2 10 ± 3 Chloroquine 89 ± 16 61 ± 9 750 ± 120 13 ± 4.6 8,9± 3,1 ngày Chlorpropamide >90 87 2,1 ± 0,4 6,8 ± 0,8 33 ± 6 Cimetidine 62 ± 6 19 540 ± 130 70 ± 14 1,9 ± 0,3 Cyclosporin 34 ± 11 96 650 ± 70 250 ± 190 16 ± 8 Diazepam 100 98,7 ± 0,2 27 ± 4 77 ± 20 43 ± 13 Digitoxin >90 97 ± 1 3,9 ± 1,3 38 ± 10 6,7± 1,7 ngày Digoxin 70 ± 13 25 ± 5 130 ± 67 640 ± 200 39 ± 13 Diltiazem 44 ± 10 78 ± 3 810 ± 130 370 ± 120 3,2 ± 1,3 Erythromycin 35 ± 25 84 ± 3 640 ± 290 55 ± 31 1,6 ± 0,7 Ethambutol 77 ± 8 20 – 30 600 ± 60 110 ± 14 3,1 ± 0,4 Furosemide 61 ± 17 98,8 ± 0,2 140 ± 30 7,7 ± 1,4 1,5 ± 0,1 Hydralazine 20 – 60 87 3900± 900 105 ± 70 1 ± 0,3 93 Liên hợp Tên thuốc SKD (F)
với protein Cl (ml/phút) Vd (l) t1/2 (h) (%) Indomethacin 98 90 140 ± 30 18 ± 5 2,4 ± 0,4 Labetalol 20 ± 5 50 1500± 630 700 ± 140 5,2 ± 1,3 Lidocaine 35 ± 11 70 ± 5 640 ± 170 77 ± 28 1,8 ± 0,4 Lithium 100 0 25 ± 8 55 ± 24 22 ± 8 Meperidine 52 ± 3 58 ± 9 1200± 350 310 ± 60 3,2 ± 0,8 Methotrexate 65 58 ± 7 110 ± 20 67 ± 14 7,2 ± 2,1 Metoprolol 38 ± 14 13 1050± 210 290 ± 50 3,2 ± 0,2 Metronidazol 99 ± 8 10 90 ± 20 77 ± 28 8,5 ± 2,9 Morphine 20 – 33 35 ± 2 1100± 140 230 ± 60 3,0 ± 1,2 Nifedipine 45 ± 28 98 720 ± 360 84 ± 35 3,4 ± 1,2 Nortriptyline 51 ± 5 92 ± 2 500 ± 130 1300 ± 300 31 ± 13 Phenobarbital 100 ± 11 51 ± 3 4,3 ± 0,9 38 ± 2 4,1 ± 0,8 ngày Procanamide 83 ± 16 16 ± 5 350 – 840 130 ± 20 3 ± 0,6 Propranolol 36 ± 10 95 ± 1 210 ± 20 42 ± 9 2,9 ± 0,8 Quinidine 80 ± 15 90 ± 3 330 ± 130 190 ± 80 6,2 ± 1,8 Ranitidine 52 ± 11 15 ± 3 730 ± 80 130 ± 20 6,2 ± 1,8 Sulfamethoxazole 100 62 ± 5 22 ± 3 15 ± 1,4 10 ± 5 Terbutaline 15 ± 6 25 210 ± 35 98 ± 28 16 ± 3 Tetracycline 77 65 ± 3 120 ± 20 105 ± 6 11 ± 1,5 Theophyllin 96 ± 8 56 ± 4 48 ± 21 35 ± 11 8,1 ± 2,4 Tolbutamide 93 ± 10 96 ± 1 21 ± 3 11 ± 2 5,9 ± 1,4 Trimethroprim 100 35 – 40 150 ± 40 130 ± 15 11 ± 1,4 Verapamil 19 ± 12 90 ± 2 830 ± 350 280 ± 60 4,8 ± 2,4 Wafarin 100 99 3,2 ± 1,7 77 ± 0,7 37 ± 15 94 PHỤ LỤC 2
Đáp án các câu hỏi lượng giá 1. Bài 1: Ca lâm sàng:
Vấn đề: Clobetasone là một thuốc steroid mạnh, được chỉ định điều trị nhiễm
nấm tại chỗ, nhưng nó có thể thấm qua da, đặc biệt ở trẻ nhỏ gây tác dụng toàn
thân. Vì trẻ sau khi bôi thuốc được quấn tả xung quanh có thể làm tăng thấm qua
da, dẫn đến hấp thụ quá mức thuốc gây tác dụng không mong muốn toàn thân là
hội chứng Cushing. Xử lý: Bác sĩ nên khuyên bà mẹ tạm dừng dùng kem
clobetasone bôi cho trẻ. Theo dõi trẻ, nếu các triệu chứng của hội chứng
Cushing dần biến mất và trẻ phát triển lại bình thường thì chứng tỏ vấn đề nghi
ngờ là đúng. Nếu không cần tìm nguyên nhân khác gây Hội chứng Cushing ở trẻ. 3. Bài 3: Ca lâm sàng
1. Khi quá liều paracetamol, con đường chuyển hóa bình thường của
paracetamol quá tải và paracetamol bị chuyển hóa qua con đường khác dưới tác
dụng của CYP1A2, CYP2E1 và CYP3A4 tạo ra một sản phẩm gây độc cao cho
gan là N-acetyl benzoquinone imine (NAPQI). Nồng độ paracetamol trong
huyết tương là 100mg/l 6 giờ sau khi uống thuốc thường không đòi hỏi điều trị
bằng thuốc giải độc, nhưng ở phụ nữ này có yếu tố nguy cơ là nghiện rượu mạn
và đang dùng carbamazepine gây cảm ứng các isoenzyme CYP2E1 và CYP3A4
nên nghiện rượu mạn và đang dùng carbamazepine và hai yếu tố nguy cơ tăng ngộ độc paracetamol.
2. N-Acetylcysteine là thuốc giải độc đặc hiệu, bởi vì nó làm thăng glutathione
giúp glutathione liên hợp với NAPQI trong tế bào gan. 5. Bài 5:
5. Phenytoin cho thấy dược động học phụ thuộc liều ; nồng độ thuốc trong huyết
tương ở liều thấp nhất 200mg/ngày cho nồng độ huyết tương là dưới khoảng liều
điều trị, vì vậy liều được tăng lên 300mg/ngày. Dù tăng tới 150% liều ban đầu,
nồng độ thuốc trong huyết tương tăng không tuyến tính (từ 25µmol/l lên
125µmol/l, gây triệu chứng ngộ độc. Nên phải giảm liều xuống 250mg/ngày thì
đạt được nồng độ thuốc trong huyết tương nằm trong khoảng liều điều trị. 95 6. Bài 6:
- Tính điểm Child-Pugh: bilirubin toàn phần = 2 điểm, albumin = 3 điểm,
prothrombin time = 3 điểm, cổ trướng = 2 điểm, bệnh não = 1 điểm. tổng số =
11 điểm. KL: rối loạn chức năng gan nặng.
- Từ Bảng 6.2, độ thanh thải theophyllin trung bình của người mắc bệnh tắc
nghẽn phổi mạn tính là 0,35 ml/phút/kg. Độ thanh thải theophyllin của trường
hợp này là: Cltheo =0,35*65= 22,8 ml/phút hoặc Cltheo = 1,37 l/h
- Liều theophyllin là: Dtheo = Css* Cltheo = 10*1,37= 14 mg/h 7. Bài 7:
1. CrCl = (Crnước tiểu*Vnước tiểu) /(SCr*T) = (50*1400) / (2,1*1440) = 23 ml/phút 2.
- Độ thanh thải creatinine:
Clcr=[0,85(140 –Tuổi)*Cân nặng / (72 *SCr) =[0,85(140 −52)65] / (72*1,8)
=37 ml/phút hoặc Clcr= (37 ml/phút) / 65 kg =0,569 ml/phút/kg
- Độ thanh thải vancomycin:
Clvan =0,695*Clcr +0,05 =0,695*0,569 +0.05 =0,446 ml/phút/kg
hoặc Clvan=0,446*65 =29 ml/phút 3.
- Độ thanh thải creatinine: Clcr=[(140 – Tuổi)*Cân nặng] / (72*SCr)
=[(140−70)*80] / (72 *2,5) =31 ml/phút
- Hằng số tốc độ thải trừ tobramycin:
ke =0,00293*Clcr +0,014 =0,00293*31 +0,014=0,105 h-1
- Thời gian bán thải của tobramycin:
t1/2=0,693/ke=0,693/0,105=6,6 h 8. Bài 8:
1. a) Sai. Carbimazole là một thuốc kháng giáp. Liều hiệu quả được xác định bằng
kiểm tra chức năng tuyến giáp thông qua theo dõi nồng độ hormon tuyến giáp.
Carbimazole nên được dừng ngay khi có dấu hiệu lâm sàng hay xét nghiệm cho
thấy giảm bạch cầu trung tính.
b) Sai. Warfarin được theo dõi thông qua INR
c) Đúng. Nồng độ đỉnh gentamicin (30 phút sau khi tiêm) tỷ lệ với hiệu quả điều
trị, nồng độ đáy tỷ lệ với độc tính của thuốc.
d) Đúng. Tăng nồng độ lithium trong huyết thanh có thể gây run, tiêu chảy, rối
loạn tiêu hóa, giảm tập trung và cuối cùng là co giật. Chức năng tuyến giáp cũng nên được theo dõi. 96
e) Đúng. Không chỉ vì ciclosporin cho thấy sự khác nhau rõ rệt giữa các cá nhân
mà sự tuân thủ điều trị cũng là một vấn đề khi điều trị cho trẻ em. Giảm chức
năng thận sau khi ghép thận có thể cho thấy hoặc là quá trình thải ghép liên quan
đến nồng độ ciclosporin thấp hoặc độc tình do tăng nồng độ thuốc quá cao.
2. Bởi vì mục tiêu cuối cùng của điều trị là không còn động kinhvà không thể
khẳng định là bệnh nhân uống đủ liều phenytoin. Một số loại động kinh xuất
hiện không thường xuyênvà có thể nồng độ phenytoin trong huyết thanh của
bệnh nhân này thấp và cô ấy chỉ đơn giản là chưa bị động kinh mà thôi. Điều
quan trọng là cần bảo đảm nồng độ thuốc nằm trong khoảng điều trị 10-20mg/l
(ở bệnh nhân với nồng độ albumin trong huyết thanh bình thường) trước khi cho
rằng bệnh nhân này đã được bảo vệ đủ khỏi cơn động kinh trong tương lai.
3. Trong trường hợp này, nên xem xét là liệu nồng độ đầu tiên được đo có đo tại
trạng thái cân bằng. Với t1/2 là 4.5h, trạng thái cân bằng hầu như đạt được sau 5
liều ((5x8h= 40 giờ) > (5t1/2 = 22,5h)). Điểm thứ hai cần xem xét là thay đổi
chức năng thận. Bởi vì aminoglycoside được đào thải chủ yếu bởi thận, giảm
chức năng thận có thể làm tăng nồng độ thuốc trong huyết thanh. Trong trường
hợp này, chức năng thận không đổi. Điểm thứ 3 cần xem xét là lỗi xét nghiệm
hoặc các chất lạ làm cản trở xét nghiệm. Nồng độ tobramycin có thể thấp "giả
tạo" nếu kháng sinh beta-lactam có thể dùng cùng một lúc; tuy nhiên trong
trường hợp này thì không. Giả thuyết lỗi labo được loại trừ, cuối cùng cần xác
định liệu có khả năng báo cáo không đúng thời điểm lấy mẫu hoặc thời điểm
dùng thuốc. Tìm hiểu kĩ mới phát hiện liều thứ 5 của tobramycin đã không
dùng. Kết quả, có thêm 8h để thuốc được loại ra khỏi cơ thể trước khi lẫy mẫu
và vì vậy nồng độ trước và sau liều thứ 6 giảm. Những nồng độ này không phản
ánh chế độ liều 100mg, mỗi 8 giờ của tobramycin. Sau khi bảo đảm là tất cả các
liều đã được tiêm đúng và rằng nồng độ các mẫu xét nghiệm lần 2 được ghi thời
điểm là đúng, điều chỉnh liều 80mg với khoảng cách 12h được chỉ định. 9. Bài 9:
1. Nồng độ toàn phần carbamazepine và acid valproic nằm trong khoảng điều trị
trong khi nồng độ phenytoin có thấp hơn nhẹ giới hạn dưới. Nếu chỉ dựa trên
nồng độ toàn phần, nếu không để ý một bác sĩ, dược sĩ có xu hướng tăng liều
hàng ngày của phenytoin lên. Nhưng khi đo nồng độ tự do các thuốc, cho thấy
bệnh nhân đã dùng liều hàng ngày carbamazepine và phenytoin phù hợp và
dùng liều quá cao acid valproic. Điều này được tái khẳng định khi các dấu hiệu
lâm sàng biến mất khi giảm liều acid valproic. Giải thích cho lý do vì sao nồng
độ acid valproic toàn phần lại thấp trong khi nồng độ tự do acid valproic lại
cao ở bệnh nhân này có thể gồm một hoặc nhiều các lí do sau: (1) phản ứng 97
gắn acid valproic với albumin có tính bão hòa (không tuyến tính) khi acid
valproic ở nồng độ cao; (2) aspirin ức chế chuyển hóa acid valproic; (3)
phenytoin và/hoặc aspirin đẩy acid valproic ra khỏi albumin. Kết quả là nồng
độ acid valproic toàn phần không phản ánh đúng với nồng độ tự do và không
thể dùng để điều chỉnh liều.
2. Khoảng điều trị đích đối với nồng độ acid valproic toàn phần được đề nghị là
50-100 mg/l. Tuy nhiên, khoảng điều trị này giả thiết nồng độ albumin nằm
trong giới hạn bình thường. Một bệnh nhân với nồng độ albumin bất thường dễ
bị độc tính khi nồng độ acid valproic toàn phần nằm trong khoảng 50-100
mg/l. Đó là bởi vì nồng độ tự do của acid valproic quá cao. Trong trường hợp
này, rất có thể là nồng độ acid valproic quá cao, dẫn đến gây lơ mơ trên bệnh
nhân. Nồng độ acid valproic toàn phần nên nằm trong khoảng dưới của khoảng
điều trị thông thường. 98
Tài liệu liên quan:
-

Basic Concepts in Medicinal Chemistry (2023) - Sách Hoá dược | Trường Đại học Y dược - Đại học Huế
33 17 -

Tổng hợp 450 Câu Trắc Nghiệm Thực vật Dược | Đại học Y dược Huế
2.9 K 1.5 K -

Đề thi Lý thuyết Dược lý II | Đại học Y dược Huế
558 279 -

Bài tập về kỹ thuật đặt Catheter tĩnh mạch cảnh trong để lọc máu cấp cứu
382 191